一种基于菲并咪唑的有机蓝光小分子及其在制备非掺杂有机电致发光器件方面的应用
1.本发明属于有机光电材料技术领域,具体涉及一种基于菲并咪唑的有机蓝光小分子及其在制备非掺杂有机电致发光器件方面的应用。
背景技术:
2.自1987年由柯达公司邓青云博士等人发明了有机发光二极管(oled)以来,在全世界范围内掀起了oled材料与器件的研究热潮。oled由于其柔性、可弯曲、广视角、节能等优点得以迅猛发展并已有相应商业化的产品应用于显示及照明领域。蓝光材料是全色显示中必不可少的三基色之一,除此之外,由于蓝光本身较宽的禁带,可以作为主体通过能量转移激发其他颜色,这不仅能使全色显示的组件制程得到简化,还能提高器件的稳定性。
3.美国电视委员会(ntsc)所定义的深蓝光的标准为蓝光坐标(0.14,0.08)。饱和的深蓝光材料不仅有利于图像显示的色彩丰富,还可以降低全色显示中的功率消耗。在商业应用领域,高效率、饱和深蓝荧光材料的紧缺已经成为制约有机电致发光器件应用的主要瓶颈之一。目前,研究比较热门的磷光材料和热活化延迟荧光(tadf)材料都能够实现100%激子利用率,但是它们不能解决蓝光目前面临的问题。对磷光材料而言,由于将辐射金属配体电荷转移(mlct)激发态提升到短波长发射区域时,金属d轨道的非辐射过程较为严重,磷光材料在深蓝区很难实现突破。对于tadf材料而言,由于激发态的强电荷转移态的特征,大多数呈现天蓝光和蓝绿光的发射,很难实现ciey≤0.08;其次,由于tadf材料涉及到长寿命的反系间窜越过程,在高电流密度下会出现严重的效率滚降,因此像磷光材料一样通常需要制备成掺杂器件,这会增加器件制备的复杂程度和经济成本,降低器件性能的可重复性。另外,由于蓝光tadf材料的最低三线态能级较高,因此对主体材料的要求也比较严苛,选择较少。因此,在ciey≤0.08的深蓝光领域并且外量子效率超过10%的非掺杂器件少之又少。为了解决这些问题,开发具有高效率、高色纯度、良好的化学和热稳定性、低效率滚降和器件制造工艺简单的蓝色有机发射器意义重大。
4.菲并咪唑类化合物可以通过“一锅法”简便的合成,原料成本低廉,修饰位点丰富,固有的双极性性质有利于载流子的传输平衡,刚性的平面有利于获得高的荧光量子产率。除此以外,菲并咪唑本身的宽禁带有利于实现深蓝光发射。将其与中性基团以及弱受体基团通过合理方式相连,构筑具有弱电荷转移特征的有机小分子材料,不仅可以实现深蓝光发射的非掺杂器件,还能实现在高亮度下的高外量子效率。
技术实现要素:
5.本发明旨在解决oleds中有机蓝光小分子材料在非掺杂器件领域色纯度低、器件效率不高、滚降严重的问题。本发明提供一种基于菲并咪唑的高效率的有机蓝光荧光小分子材料,这类材料合成方法简单,热稳定性出色。该类化合物应用于电致发光领域实现了高效率深蓝光发射,高亮度下的低滚降等优秀特性,克服了金属配合物磷光材料和tadf材料
必须要掺杂且高亮度下器件效率衰减严重等缺点。
6.为实现上述目的,本发明所述的一种基于菲并咪唑的有机蓝光小分子,其结构通式如下所示:
[0007][0008]
取代基r1-r4可以相同也可以不同,选自氢、苯基、联苯基、三苯基、氰基取代苯基、氟苯基、二氟苯基、三氟甲基苯基、萘基、蒽基、二苯醚基、二苯硫醚基、二苯基甲烷基、芴基及芴基的衍生物、螺芴基及螺芴基衍生物等其他芳香结构。
[0009]
优选的,本发明所述的一种基于菲并咪唑的有机蓝光小分子,其结构式如下之一所示:
[0010][0011]
上述基于菲并咪唑的有机蓝光小分子,其是从菲醌出发一锅法制备菲并咪唑原料及利用suzuki偶联制备得到目标化合物。
[0012]
一种基于上述有机蓝光小分子制备的非掺杂有机电致发光器件,由玻璃基板、ito阳极、空穴传输层、发光层、电子传输层和阴极组成,其特征在于:发光层至少含有一种本发明所述的有机蓝光小分子。
[0013]
本发明的原理为:菲并咪唑具有大的刚性π平面,有高的荧光量子产率,同时其本
身的宽禁带有助于实现其衍生物的深蓝光发射。菲并咪唑的双极性特征有助于载流子的传输平衡,将中性基团与菲并咪唑相连接不会引起强的分子内电荷转移,有利于构筑弱电荷转移特征的分子,使目标产物的光色能够蓝移到短波长区。除此以外,选用具有一定空间位阻的基团,增大分子间的间距,避免分子固态下的聚集淬灭现象,实现高的固态发光效率,为实现高效非掺杂蓝光电致发光器件奠定了基础。
[0014]
本发明所述的有机蓝光小分子发光材料及由其制备的非掺杂有机电致发光器件具有如下的特点:
[0015]
1、制备方法简单,反应条件温和,制备的目标产物具有优异的热稳定性和化学稳定性。
[0016]
2、本发明基于菲并咪唑的衍生物具有合适的能级,有利于载流子的传输和平衡,可作为蓝光材料应用于有机电致发光领域。
[0017]
3、本发明的衍生物具备分子内弱电荷转移特性,可以实现饱和蓝光发射,推动了蓝色电致发光材料的进一步发展。
[0018]
4、本发明的有机小分子适合制备非掺杂蓝光oled器件,器件结构更加简单,节约制作成本。
附图说明
[0019]
图1是p2的热失重曲线,热分解温度(td)为430℃,具有比较好的热稳定性,为真空蒸镀制备发光器件提供了基础。
[0020]
图2是p2的示差扫描量热曲线,玻璃化转变温度(tg)为134℃,结晶峰(tc)为225℃,熔点(tm)为302℃;如此高的结晶和熔点温度保障其在oled器件工作时的形态学稳定性,可以有效提升器件效率的稳定性和使用寿命。
[0021]
图3是p2非掺杂蒸镀薄膜的吸收和发射光谱,吸收主峰分别位于334nm、和369nm,发射主峰位于440nm,为蓝光的发射,可以作为有机蓝光材料制备电致发光器件。
[0022]
图4是以化合物p1为发光层制备的非掺杂电致发光器件的亮度-电压-电流密度曲线,器件的最大亮度为5634cd m-2
,开启电压为3.3v;
[0023]
图5是以化合物p1为发光层制备的非掺杂电致发光器件的电流效率-亮度-能量效率曲线,最大的电流效率为2.6cd a-1
,最大的功率效率为2.0lm w-1
;
[0024]
图6是以化合物p1为发光层制备的非掺杂电致发光器件的外量子效率曲线,最大的外量子效率为6.1%:插图:亮度为100cd m-2
时的电致发光光谱,光谱主峰位于424nm;
[0025]
图7是以化合物p1为发光层制备的非掺杂电致发光器件在不同电压下的电致发光光谱,光谱主峰位于424nm,电致发光光谱在不同驱动电压下的重复度高说明稳定性好。
[0026]
图8是以化合物p2为发光层制备的非掺杂电致发光器件的亮度-电压-电流密度曲线,器件的最大亮度为18637cd m-2
,开启电压为3.0v;
[0027]
图9是以化合物p2为发光层制备的非掺杂电致发光器件的电流效率-亮度-能量效率曲线,最大的电流效率为7.2cd a-1
,最大的功率效率为6.2lm w-1
;
[0028]
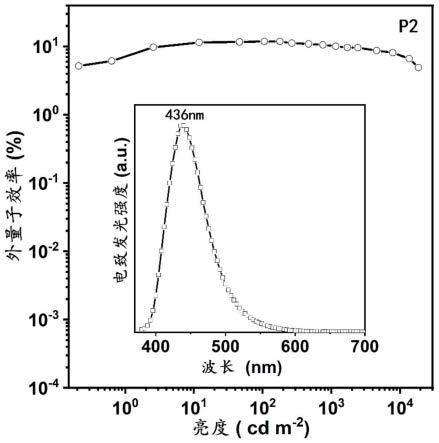
图10是以化合物p2为发光层制备的非掺杂电致发光器件的外量子效率曲线,最大的外量子效率为11.8%:插图:亮度为100cd m-2
的电致发光光谱,光谱主峰位于436nm;
[0029]
图11是以化合物p2为发光层制备的非掺杂电致发光器件在不同电压下的电致发
光光谱,光谱主峰位于436nm,电致发光光谱在不同的驱动电压下是稳定的。
具体实施方式
[0030]
下面结合附图和实施例对本发明作进一步描述,以便于所属技术领域的人员对本发明的理解。显然,所描述的实施例仅仅是本实验的一部分,并不是全部的实施例,所属领域的技术熟练人员,根据上述发明内容对本发明做出的非本质性修改、等同替换和改进等,均应包含在本发明的保护范围之内。下述所提及的原料均为市售或者按照已知文献或专利进行制备,未提及的工艺步骤和制备方法均为本领域技术人员所熟知的工艺步骤和制备方法。
[0031]
实施例1:本实施例化合物p1的制备,其步骤如下:
[0032][0033]
m1的合成:m1通过一锅法合成,100ml圆底烧瓶中,将菲醌(2.08g,10.00mmol)、苯胺(3.73g,40.00mmol)、3,5-二溴苯甲醛(2.64g,10.00mmol)和醋酸铵(3.85g,50mmol)溶于50ml冰醋酸溶液,120℃加热回流2小时。反应结束后用水淬灭反应,抽滤得到固体,依次用水、冰醋酸、乙醇洗,得到粗产品,柱层析分离提纯(石油醚:二氯甲烷体积比=2:1),得到白色固体(4.23g,产率:80%)。质谱maldi-tof(m/z)[m
+
]:测量值为528.77,理论值为528.25。
[0034][0035]
m2的合成:将m1(5.28g,10.00mmol)、联硼酸频那醇酯(10.12g,40.00mmol)、乙酸钾(5.88g,60mmol)和[1,1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(490mg,0.6mmol)溶于80ml 1,4-二氧六环溶液,85℃加热回流48小时。反应结束后用二氯萃取分液,浓缩得到粗产品,柱层析分离提纯(石油醚:二氯甲烷体积比=2:3),得到白色固体(3.73g,产率:60%)。质谱maldi-tof(m/z)[m
+
]:测量值为622.88,理论值为622.38。
[0036][0037]
p1的合成:100ml圆底烧瓶,将m2(1.87g,3.0mmol)、溴苯(1.13g,7.2mmol)、碳酸钾(5.53g,40mmol)、四(三苯基膦)钯(187.20mg,0.18mmol)溶于20ml蒸馏水和40ml甲苯中,氮
气保护下90℃回流24小时。用二氯甲烷萃取分液,浓缩得到粗产品,柱层析分离提纯(石油醚:二氯甲烷体积比=4:1),得到白色固体(1.25g,产率:80%)。质谱:maldi-tof(m/z)[m
+
]:测量值为522.45,理论值为522.65;元素分析:c
39h26
n2理论值为c 89.63,h 5.01,n 5.36;测量值为c 89.61,h 5.03,n 5.36。
[0038]
实施例2:本实施例化合物p2的制备,其步骤如下:
[0039][0040]
m3的合成:m3通过一锅法合成,100ml圆底烧瓶中,将菲醌(2.08g,10.00mmol)、苯胺(3.73g,40.00mmol)、4-溴苯甲醛(1.85g,10.00mmol)和醋酸铵(3.85g,50mmol)溶于50ml冰醋酸溶液,120℃加热回流2小时。反应结束后用水淬灭反应,抽滤得到固体,依次用水、冰醋酸、乙醇洗,得到粗产品,柱层析分离提纯(石油醚:二氯甲烷体积比=2:1),得到白色固体(3.59g,产率:80%)。质谱maldi-tof(m/z)[m
+
]:测量值为449.55,理论值为449.35。
[0041][0042]
m4的合成:将m3(4.49g,10.00mmol)、联硼酸频那醇酯(5.06g,20.00mmol)、乙酸钾(2.94g,30mmol)和[1,1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(245mg,0.3mmol)溶于80ml 1,4-二氧六环溶液,85℃加热回流48小时。反应结束后用二氯萃取分液,浓缩得到粗产品,柱层析分离提纯(石油醚:二氯甲烷体积比=2:3),得到白色固体(3.48g,产率70%)。质谱maldi-tof(m/z)[m
+
]:测量值为496.39,理论值为496.42。
[0043][0044]
p2的合成:100ml圆底烧瓶,将m4(1.49g,3.0mmol)、5'-溴间三联苯(1.11g,3.6mmol)、碳酸钾(5.53g,40mmol)、四(三苯基膦)钯(93.60mg,0.09mmol)溶于20ml蒸馏水和40ml甲苯中,氮气保护下90℃回流24小时。用二氯甲烷萃取分液,浓缩得到粗产品,柱层析分离提纯(石油醚:二氯甲烷体积比=4:1),得到白色固体(1.25g,产率:80%)。质谱:maldi-tof(m/z)[m
+
]:测量值为598.24,理论值为598.75;元素分析:c
45h30
n2理论值为c 90.27,h 5.05,n 4.68;测量值为c 90.29,h 5.04,n 4.67。
[0045]
实施例3:本实施例化合物p3的制备,其步骤如下:
[0046][0047]
本实施例与实施例2基本相同,其不同之处在于:本例中需将5'-溴间三联苯换为等物质的量的4-溴-5'-苯基-1,1':3',1
”‑
三联苯,得到白色粉末状固体(1.35g,产率:80%)。质谱:maldi-tof(m/z)[m
+
]:测量值为674.27,理论值为674.85;元素分析:c
51h34
n2理论值为c 90.77,h 5.08,n 4.15;测量值为c 90.73,h 5.09,n 4.17。
[0048]
实施例4:本实施例化合物p4的制备,其步骤如下:
[0049][0050]
本实施例与实施例2基本相同,其不同之处在于:本例中需将5'-溴间三联苯换为等物质的量的3-溴-5'-苯基-1,1':3',1
”‑
三联苯,得到白色粉末状固体(1.35g,产率:80%)。质谱:maldi-tof(m/z)[m
+
]:测量值为674.27,理论值为675.05;元素分析:c
51h34
n2理论值为c 90.77,h 5.08,n 4.15;测量值为c 90.73,h 5.09,n 4.17。
[0051]
实施例5:本实施例化合物p5的制备,其步骤如下:
[0052][0053]
本实施例与实施例1基本相同,其不同之处在于:本例中需将溴苯换为等物质的量的5'-溴间三联苯,得到白色粉末状固体(1.73g,产率:70%)。质谱:maldi-tof(m/z)[m
+
]:测量值为826.33,理论值为827.04;元素分析:c
63h42
n2理论值为c 91.49,h 5.12,n 3.39;测量值为c 91.46,h 5.14,n 3.40。
[0054]
实施例6:本实施例化合物p6的制备,其步骤如下:
[0055][0056]
本实施例与实施例2基本相同,其不同之处在于:本例中需将5'-溴间三联苯换为等物质的量的4-溴代联苯,得到白色粉末状固体(1.25g,产率:80%)。质谱:maldi-tof(m/z)[m
+
]:测量值为522.21,理论值为522.65;元素分析:c
39h26
n2理论值为c 89.63,h 5.01,n 5.36;测量值为c 89.64,h 5.02,n 5.34。
[0057]
实施例7:本实施例化合物p7的制备,其步骤如下:
[0058][0059]
本实施例与实施例2基本相同,其不同之处在于:本例中需将5'-溴间三联苯换为等物质的量的间溴联苯,得到白色粉末状固体(1.25g,产率:80%)。质谱:maldi-tof(m/z)[m
+
]:测量值为522.21,理论值为522.65;元素分析:c
39h26
n2理论值为c 89.63,h 5.01,n 5.36;测量值为c 89.64,h 5.02,n 5.34。
[0060]
实施例8:本实施例化合物p8的制备,其步骤如下:
[0061][0062]
本实施例与实施例2基本相同,其不同之处在于:本例中需将5'-溴间三联苯换为等物质的量的4-溴联苯醚,得到白色粉末状固体(1.29g,产率:80%)。质谱:maldi-tof(m/z)[m
+
]:测量值为538.20,理论值为538.65;元素分析:c
39h26
n2o理论值为c 89.96,h 4.87,n 5.20;测量值为c 89.94,h 4.89,n 5.20。
[0063]
实施例9:本实施例化合物p9的制备,其步骤如下:
[0064][0065]
本实施例与实施例2基本相同,其不同之处在于:本例中需将5'-溴间三联苯换为等物质的量的4-溴二苯基甲烷,得到白色粉末状固体(1.29g,产率:80%)。质谱:maldi-tof(m/z)[m
+
]:测量值为536.23,理论值为536.68;元素分析:c
40h28
n2理论值为c 89.52,h 5.26,n 5.22;测量值为c 89.54,h 5.28,n 5.20。
[0066]
实施例10:本实施例化合物p10的制备,其步骤如下:
[0067][0068]
本实施例与实施例2基本相同,其不同之处在于:本例中需将5'-溴间三联苯换为等物质的量的2-溴-9,9-二甲基芴,得到白色粉末状固体(1.35g,产率:80%)。质谱:maldi-tof(m/z)[m
+
]:测量值为562.25,理论值为563.75;元素分析:c
42h30
n2理论值为c 89.65,h 5.37,n 4.98;测量值为c 89.67,h 5.39,n 4.94。
[0069]
实施例11:本实施例化合物p11的制备,其步骤如下:
[0070][0071]
本实施例与实施例2基本相同,其不同之处在于:本例中需将5'-溴间三联苯换为等物质的量的2-溴-9,9-二苯基芴,得到白色粉末状固体(1.65g,产率:80%)。质谱:maldi-tof(m/z)[m
+
]:测量值为686.72,理论值为686.68;元素分析:c
52h34
n2理论值为c 90.93,h 4.99,n 4.08;测量值为c 90.91,h 4.99,n 4.10。
[0072]
实施例12:本实施例化合物p12的制备,其步骤如下:
[0073][0074]
本实施例与实施例2基本相同,其不同之处在于:本例中需将5'-溴间三联苯换为等物质的量的2-溴-9,9-螺二芴,得到白色粉末状固体(1.64g,产率:80%)。质谱:maldi-tof(m/z)[m
+
]:测量值为684.26,理论值为684.84;元素分析:c
52h32
n2理论值为c 91.20,h 4.71,n 4.09;测量值为c 91.19,h 4.73,n 4.08。
[0075]
实施例13:本实施例化合物p13的制备,其步骤如下:
[0076][0077]
本实施例与实施例2基本相同,其不同之处在于:本例中需将5'-溴间三联苯换为等物质的量的对溴苯腈,得到浅绿色粉末状固体(1.13g,产率:80%)。质谱:maldi-tof(m/z)[m
+
]:测量值为471.17,理论值为471.56;元素分析:c
34h21
n3理论值为c 86.60,h 4.49,n 8.91;测量值为c 86.58,h 4.50,n 8.92。
[0078]
实施例14:本实施例化合物p14的制备,其步骤如下:
[0079][0080]
本实施例与实施例2基本相同,其不同之处在于:本例中需将5'-溴间三联苯换为等物质的量的1-溴-3,5-二氟苯,得到白色粉末状固体(1.16g,产率:80%)。质谱:maldi-tof(m/z)[m
+
]:测量值为482.16,理论值为482.53;元素分析:c
33h20
f2n2理论值为c 82.14,h 4.18,n 5.81;测量值为c 82.12,h 4.16,n 5.82。
[0081]
实施例15:本实施例化合物p15的制备,其步骤如下:
[0082][0083]
本实施例与实施例2基本相同,其不同之处在于:本例中需将5'-溴间三联苯换为等物质的量的对溴三氟甲苯,得到白色粉末状固体(1.16g,产率:80%)。质谱:maldi-tof(m/z)[m
+
]:测量值为514.17,理论值为514.55;元素分析:c
34h21
f3n2理论值为c 79.37,h 4.11,n 5.44;测量值为c 79.35,h 4.12,n 5.45。
[0084]
效果实施例1~15
[0085]
以下利用本发明的材料p1~p15制备非掺杂电致发光器件实施例,具体的器件制备工艺及器件性能测试实验操作如下:基片铟锡氧化物(ito)导电玻璃的准备:基片依次用去离子水、异丙醇、丙酮、甲苯、丙酮、异丙醇在超声浴中各自清洗20分钟,并于烘箱中烘干备用。在紫外臭氧清洗机中对ito导电玻璃表面处理40分钟后,将其移入真空蒸镀设备中(腔体内压力<2
×
10-4
pa);在阳极ito导电玻璃上,真空蒸镀空穴注入层hatcn,厚度为6nm;在hatcn上,真空蒸镀空穴传输层tapc,厚度为25nm:在tapc上,真空蒸镀激子阻挡层tcta,厚度为15nm;在tcta上,真空蒸镀发光层,厚度为20nm;在p1上,真空蒸镀电子传输层tmpypb,厚度为40nm;在tmpypb上,真空蒸镀电子注入层lif,厚度为1nm;在lif上,真空蒸镀阴极al,厚度为120nm。
[0086]
所述有机电致发光非掺器件的结构如下:ito/hatcn(6nm)/tapc(25nm)/tcta(15nm)/eml(20nm)/tmpypb(40nm)/lif(1nm)/al(120nm),eml代表发光层,发光层为本发明的材料真空蒸镀的非掺杂薄膜。
[0087]
针对本实施例制备得到的有机电致发光器件施加直流电压,使用spectrascan pr655亮度计来评价发光性能,使用电脑控制的keithley 2400数字源表测量电流-电压特性。作为发光特性,测定在随外加直流电压变化下的开启电压、最大亮度、发光峰位、cie色坐标值、外量子效率等参数,效果实施例结果见表1~表3。
[0088]
表1:效果实施例1器件的电致发光性能数据
[0089][0090]
表2:效果实施例2器件的电致发光性能数据
[0091][0092]
表3:效果实施例1~15器件的电致发光性能数据
[0093][0094][0095]
效果实施例1-15的有机电致发光器件中所用的材料结构式如下,均可购买得到:
[0096][0097]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1