一种快速低丰度检测插入和缺失突变的方法
1.本发明涉及核酸序列检测领域,具体涉及dna序列的识别,插入和缺失突变的分型以及插入和缺失突变检测等技术领域。更具体地说它是一种快速低丰度检测插入和缺失突变的检测方法。更具体第说它是一种基于cas 12a酶和rpa技术联用的快速低丰度检测插入和缺失突变的方法。
背景技术:
2.基因突变与多张疾病密切相关,基因突变包括插入、缺失和点突变等。尽管插入和缺失突变在疾病中出现的频率不高,但是其对疾病的发生发展具有重要的作用。例如,肺癌的egfr基因外显子19缺失突变,已经作为普查肺癌的依据之一。因此,插入和缺失突变检测对于疾病的诊断和治疗具有重要的指导意义。传统的插入和缺失突变检测主要是通过组织活检来获取样本,然后提取组织dna进行后续分析,这种侵入式操作不仅对病人造成的痛苦较大,而且对操作的器材和人员要求高,不适合连续多次采样和动态监测。此外,组织活检技术依赖影像学可见的实体病灶进行定位,难以实现早期诊断。有别于组织活检,新兴的液体活检技术主要通过分析体液尤其是血液样本中的胞外游离dna来反映机体的基因突变情况。此方法操作简单,成本低,可以连续动态地提供基因突变信息,具有广阔的应用前景。
3.基因检测方法主要包括:选择性pcr、数字pcr、测序和核酸探针技术。然而,选择性pcr、数字pcr、测序这三种检测技术均无法兼具简便快速、高灵敏度、低成本的检测能力,进一步改进上述技术,所需的研发投入大,周期长。因此,需要另辟蹊径。
4.选择性pcr以阻滞剂pcr(blocker pcr),低温变性共扩增pcr(cold-pcr)为代表,可以实现对丰度0.1%样品的检测,仅仅部分满足突变检测的灵敏度要求。数字pcr技术可分为芯片式和液滴式两种形式,基本原理都是将大量稀释的dna样品液分散至微容器或微液滴,通过后续的扩增反映,实现目标dna的定量检测。数字pcr可以检测丰度低至0.01%的样品,但是数字pcr仪器昂贵、操作复杂。目前,基于测序的癌症基因检测主要使用的是高通量测序,它极高的多重检测能力不仅能够提供全面的基因突变信息,而且具备筛选未知突变的能力,在寻找致病基因和癌症药物基因组学方面有着不可替代的优势。但是,整个测序过程的持续时间长,花费高。因此,对于临床意义明确的已知致病缺失或插入突变,使用高通量测序技术进行检测的性价比较低,不适合大规模推广应用。
5.核酸探针技术需要对每个突变位点设计特异性的探针。这不仅意味着需要合成多种探针增加了检测成本,而且优化过程中不断调整探针序列会大幅增加优化的难度和成本,并使多重检测更加困难。而基于cas12a核酸酶具有速度更快,信号更强的优势,近年来基于cas12a核酸酶的检测体系得到了较好的发展,但是目前也无法直接检测组织或血浆中提取出的dna。基于cas12a核酸酶的检测体系常常与pcr技术联用,都是pcr后检测,而pcr是一个需要耗费时间很长,且操作复杂的过程。
6.近年来,核酸探针技术在癌症基因检测中取得了快速发展和广泛应用,其具有快速、高灵敏等特点,有望实现突破。根据是否将探针整合进pcr过程,核酸探针技术可分为实
时检测和post-pcr检测。在post-pcr检测模式中,探针的设计无需考虑复杂的pcr过程,因而可以充分发挥dna结构的可塑性,并挖掘其与核酸酶的相互作用,这些基于dna探针与核酸酶相互作用的检测技术具有很高的灵敏度,检测限可达0.01%。在这些体系中,核酸酶往往通过酶切探针/目标链复合物来产生信号的同时,释放目标链去结合下一个探针,从而实现底物链的循环利用,放大其信号。核酸酶信号放大的能力越强,检测体系所需的目标链浓度就越低,这不仅可以大幅降低对前期pcr扩增的要求,也更容易检测到更低丰度的突变。因此,优化dna探针的结构和序列设计并寻找更强大的信号放大核酸酶是核酸探针技术发展的重要方向。
7.与大部分核酸酶的信号放大机制不同,cas12a不再通过“目标链循环多次反应”来放大信号,而是在cas12a/grna复合物结合目标链之后激活cas12a,被激活的cas12a具有核酸内切酶活性(dnase活性),可以不断的酶切溶液中大量存在的荧光dna探针,实现信号的放大。这一信号放大过程不涉及到循环往复的dna结合与解离,因而速度更快,信号更强。此外,体系中荧光dna探针的序列与目标突变的序列可以完全无关,因此检测不同的目标突变时,无需更换探针,有利于构建突变检测体系。鉴于cas12a优异的信号放大能力和探针的通用性,基于crispr-cas12a的核酸探针体系被广泛研究并不断优化。然而,目前核酸酶检测体系常常与pcr技术联用,而pcr是一个需要耗费时间很长,且操作复杂的过程,大大影响了核酸酶探针检测体系的临床应用。重组酶聚合酶扩增(recombinase polymerase amplification,rpa),被称为是可以替代pcr的核酸检测技术。rpa技术可大大缩短常规pcr扩增所需的时间,从而缩短检测所需的时间。因此,在基因检测方面,还没有开发出快速低丰度检测插入和缺失突变的检测方法。基于cas12a的核酸探针技术具有极强的信号放大功能,在与简单快速的恒温扩增技术联用,有望实现高灵敏度、低成本、简便快速的插入和缺失突变检测。
8.因此,开发一种高灵敏度、低成本的快速低丰度检测插入和缺失突变的检测方法很有必要。
技术实现要素:
9.本发明的目的是为了提供一种基于cas 12a酶和rpa技术联用的快速低丰度检测插入和缺失突变的方法,成本低,检测速度快,灵敏度高,本发明方法能够检测到极微量的目标基因序列,且由于crispr-cas12a技术的靶向性极佳,能够检测出极低突变丰度的插入或缺失基因。
10.为了实现上述目的,本发明的技术方案为:一种快速低丰度检测插入和缺失突变的方法,其特征在于:包括如下步骤,
11.步骤一:制备双标记荧光探针,
12.探针为单链dna,一端标记荧光基团,另一端标记猝灭基团,探针长度设计为大于或等于6个核苷酸残基,te buffer稀释液稀释探针粉末,配置成液体,用nanodrop仪器测定浓度至浓度20um,配置成5um的液体,待用;
13.步骤二:制备cas12a(cpf1)核酸酶,浓度为2000pmol,使用超纯水稀释100倍,待用;
14.步骤三:挑选临床上最为常见的突变基因作为验证靶点,在pubmed网站上查询缺
失突变点基的缺失突变点序列,
15.突变基因包括缺失突变基因、插入突变基因;缺失突变基因包括缺失突变egfr外显子19(-ggaattaagagaagc)、缺失突变cd41-42(-cttt);
16.插入突变基因包括插入突变egfr20ins;
17.依据实验原理分别设计所需的突变链,野生链,grna;
18.步骤四:用te buffer稀释液稀释步骤二所设计合成的链粉末,配置成液体,用nanodrop仪器测定浓度至20um,配置成2um的野生链和突变链的双链dna液体;
19.在配置成野生链和突变链的双链dna液体时:通过升温退火的方法制备成双链,升温85
°
,退火55
°
,置于37
°
下10分钟;
20.步骤五:配制cas12a/grna混合反应液;
21.配制cas12a/grna混合反应液:1.1倍的2.1buffer(10x),50nm的grna,75nm的cas12a,超纯水补齐,待用;
22.步骤六:将步骤四中的液体配制成双链的野生链和突变链,浓度为0.5um,待用;
23.配制成双链的野生链和突变链的方法为:通过升温退火的方法制备成双链,升温85
°
,退火55
°
,置于37
°
下10分钟;
24.步骤七:配制50ul的待检反应体系:5ul的2.1buffer,5ul的步骤五待用液,2ul的步骤六待用液,2ul的mgso4(10mmol),2ul的步骤一待用液,34ul超纯水;
25.步骤八:将步骤七的待检反应体系放置于仪器中检测荧光曲线,读取数据;
26.步骤九:分别使用突变基因的基因缺失突变的合成野生链来稀释突变链,制备一系列浓度为0.5um混合样本,使其突变丰度范围为0.1%、0.01%、0.001%、0%;目的是为了探究本方法可以检测的突变丰度,也就是检测限;
27.步骤十:配制50ul的待检反应体系:5ul的2.1buffer,5ul的步骤五待用液,2ul的步骤九待用液,2ul的mgso4(10mmol),2ul的步骤一待用液,34ul超纯水;
28.步骤十一:将步骤十的待检反应体系放置于仪器中检测荧光曲线,读取数据;
29.步骤十二:依据合成的突变基因的突变链和野生链的核苷酸残基序列,设计rpa反应的引物;
30.步骤十三:用te buffer稀释液稀释步骤十二所设计合成的链粉末,配置成液体,用nanodrop仪器测定浓度至50um,配置成10um的液体;
31.步骤十四:配置rpa反应体系:分别向试剂盒的rpa反应管中加入稀释2000倍的步骤九的0.5um的0.1%、0.01%、0.001%、0%混合液体,2ul的rpa反应的引物fp和rp至于管盖,此步骤为冰上操作;盖上管盖,离心,离心管离心速度为1000转/min;37℃条件下反应10分钟;
32.步骤十五:配制50ul的待检反应体系:5ul的2.1buffer,5ul的步骤五待用液,20ul的步骤十四待用液,2ul的mgso4(10mmol),2ul的步骤一待用液,16ul超纯水;
33.步骤十六:将步骤十五的待检反应体系放置于仪器中检测荧光曲线,读取数据。
34.在上述技术方案中,当突变基因选用缺失序列egfr外显子19(-ggaattaagagaagc)时,
35.突变链,野生链,grna的设计原理:突变链和野生链,设计包含缺失序列egfr外显子19(-ggaattaagagaagc)及其缺失序列的3
‘
端和5
‘
端若干个碱基,突变链和野生链的总长
度为100-130个碱基,缺失序列位于整个序列的相对中间位置。
36.设计的grna针对缺失的突变链序列设计,grna是靶向突变链;在egfr外显子19(-ggaattaagagaagc)缺失突变的原始序列3
‘
端和5
‘
端设计grna,分别在3
‘
端和5
‘
端设计9-10个碱基,即grna序列与突变链匹配的碱基数为18-20个碱基,总长度为40-42个碱基。
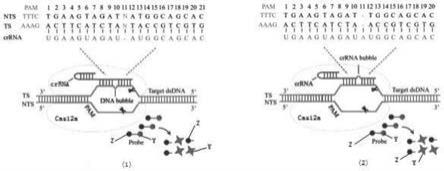
37.在上述技术方案中,在步骤三中,当突变基因选用缺失序列egfr外显子19(-ggaattaagagaagc)时,突变链的长度为106核苷酸残基,野生链的长度为121核苷酸残基,grna长度为40个核苷酸残基。
38.在上述技术方案中,在步骤三中,当突变基因选用缺失序列egfr外显子19(-ggaattaagagaagc)时,突变链的长度为106核苷酸残基,序列为:
39.gggactctggatcccagaaggtgagaaagttaaaattcccgtcgctatcaaaacatctccgaaagccaacaaggaaatcctcgatgtgagtttctgctttgctgtg,其互补序列为cacagcaaagcagaaactcacatcgaggatttccttgttggctttcggagatgttttgatagcgacgggaattttaactttctcaccttctgggatccagagtccc,
40.野生链的长度为121核苷酸残基,序列为:
41.gggactctggatcccagaaggtgagaaagttaaaattcccgtcgctatcaaggaattaagagaagcaacatctccgaaagccaacaaggaaatcctcgatgtgagtttctgctttgctgtg,其互补序列为cac agc aaa gca gaa act cac atc gag gat ttc ctt gtt ggc ttt cgg aga tgt tgc ttc tct taa ttc ctt gat agc gac ggg aat ttt aac ttt ctc acc ttc tgg gat cca gag tcc c,
42.grna的长度为40个核苷酸残基,序列为:
43.uaauuucuacuaaguguagauggagauguuuugauagcga。
44.在上述技术方案中,在步骤十二中,rpa反应的引物的设计原理为:
45.扩增长度在100-130bp之间,引物长30-35个碱基,不使用聚嘌呤和嘧啶,且gc含量在30-70%之间。
46.在上述技术方案中,当突变基因选用缺失序列egfr外显子19(-ggaattaagagaagc)时,依据合成的egfr外显子19(-ggaattaagagaagc)基因突变链的长度为106和野生链的长度为121的核苷酸残基序列,设计rpa反应的引物,
47.引物序列为fp:gggactctggatcccagaaggtgagaaag,
48.rp:cacagcaaagcagaaactcacatcgaggatttc。
49.在上述技术方案中,在步骤五中,配制25ul的cas12a/grna混合反应液:2.8ul的2.1buffer,2.2ul的步骤二待用液,0.94ul grna(1um)和19.06的超纯水,待用。
50.在上述技术方案中,在步骤一中,探针的长度一般设置为6-8个核苷酸残基;探针的序列为ta碱基交错排列。
51.在上述技术方案中,在步骤一中,探针长度设计为8核苷酸残基,探针的序列:ttattatt。
52.在上述技术方案中,在步骤一中,荧光基团及其猝灭基团应当满足荧光基团的发射光谱与猝灭基团的吸收光谱有效重叠;
53.荧光基团及其猝灭基团至少选自fam与tamra,fam与bhq1,tet与bhq2。
54.本发明具有如下优点:
55.(1)本发明方法通过基于cas 12a核酸酶和rpa技术联用检测插入和缺失基因突
变,通过cas 12a核酸酶特异性靶向切割目标链识别基因突变序列,通过cas 12a核酸酶非特异性切割活性无限方法目标信号,通过核酸酶高灵敏度实现低丰度检测,通过rpa技术大大缩短常规pcr扩增所需的时间,达到快速低丰度检测插入和缺失突变;本方法在多重插入和缺失基因突变检测方面具有极大的优势,在未来临床应用中具有广泛的前景。
56.(2)本发明中的cas12a酶的高效酶切活性,检测灵敏度高(本发明的灵敏度可以达到0.001%);
57.(3)本发明中的探针具有通用性,换其他的插入或缺失突变位点检测,探针不用换,检测成本低(本发明的检测成本约几十块钱;而现有技术如测序或者ddpcr,其检测成本几百到几千不等);
58.(4)本发明结合rpa扩增技术以及cas12a酶的高效酶切活性,大大缩短了检测时间,检测速度快(本发明的检测耗时为2.5小时及以内;而现有技术如测序或者ddpcr,由于其检测过程繁琐,检测耗时为一两天至一周左右)。
附图说明
59.图1为本发明中的目标序列单碱基插入、缺失情况模拟图。
60.图2为本发明中的目标序列单碱基插入、缺失情况检测。
61.图3为本发明egfr外显子19缺失突变的检测图。
62.图4为本发明egfr外显子19缺失突变的检测下线。
63.图5为本发明rpa反应扩增egfr外显子19缺失突变基因流程图。
64.图6为本发明cas12a和rpa反应联用检测egfr外显子19缺失突变图。
65.图7为现有技术通用型探针熔解曲线法的egfr外显子19缺失突变位点的荧光检测曲线。
66.在图1中,图1(1)表示目标序列单碱基插入情况模拟图;图1(2)表示目标序列单碱基缺失情况模拟图。在图1(1)和图1(2)中,y表示荧光基团;z表示猝灭基团。
67.在图2中,图2(1)表示目标序列单碱基插入情况的检测图;图2(2)表示目标序列单碱基缺失情况检测图。在图2(1)和图2(2)中,c表示c碱基,g表示g碱基,a表示a碱基,t表示t碱基。
68.在图3中,mt为突变,wt为正常;在图3中,横坐标表示时间,单位s;纵坐标表示荧光强度。
69.在图4中,突变丰度分别为0.1%、0.01%、0.001%、0%。在图4中,横坐标表示时间,单位s;纵坐标表示荧光强度。
70.在图6中,突变丰度为0.1%、0.01%、0.001%、0%。在图6中,横坐标表示时间,单位s;纵坐标表示荧光强度。
71.在图4、图6中,a、b、c、d分别表示不同的曲线,曲线表示不同的突变丰度。
72.在图7中,a、b、c、d、e、f分别表示不同的曲线,曲线表示不同的突变丰度。在图7中,横坐标表示温度,单位℃;纵坐标表示衍生物。
具体实施方式
73.下面结合附图详细说明本发明的实施情况,但它们并不构成对本发明的限定,仅
作举例而已。同时通过说明使本发明的优点更加清楚和容易理解。
74.本发明方法首先利用等温扩增技术(rpa),快速扩增出大量的待测基因组中的目标基因序列,再利用设计好的crispr-cas12a技术,它能够特异性的识别目标基因序列,进而cas12a酶被激活。激活后的cas12a酶会切割反应体系中的核酸探针,当探针被切割断裂之后,fam基团和bhq基团就会分离,然后产生荧光。本发明通过cas 12a核酸酶特异性靶向切割目标链识别基因突变序列,通过cas 12a核酸酶非特异性切割活性无限方法目标信号,通过核酸酶高灵敏度实现低丰度检测,通过rpa技术大大缩短常规pcr扩增所需的时间,达到快速低丰度检测插入和缺失突变。
75.本发明采用的原理是:第一部分:为了确定crispr-cas12a系统是否能够很好的检测插入突变,本发明随机设计序列作为模型,合成与靶序列匹配长度为20-nt的grna-1。在grna-1的20-nt靶序列中,系统地在所有可能的位置添加核苷酸,在目标靶序列中形成碱基插入,模拟碱基插入情况(如图1(1)所示)。此外,为了确定crispr-cas12a系统是否能够很好的检测缺失突变,在grna-1的20-nt靶序列中,系统地在所有可能的位置删除核苷酸,在grna序列中形成碱基缺失,模拟碱基缺失情况(如图1(2)所示)。此部分证实crispr-cas12a系统能够很好的检测插入和缺失突变,结果可以看出在不同的位置插入或缺失突变的检测效果也是不同的(如图2所示)。插入突变的检测效果如图2(1)所示,缺失突变的检测效果如图2(2)所示。然后,以实际致病缺失突变egfr外显子19,简称19del为例,设计基因的合成序列,以及合成18-nt grna-2。此部分证实crispr-cas12a系统能够很好的检测egfr外显子19(如图3所示)。同时在合成的人工序列中,检测了本发明方法对egfr外显子19的检测下线(如图4所示)。第二部分:设计egfr外显子19缺失突变的rpa引物,通过rpa反应扩增得到所需的egfr外显子19突变检测基因序列,其中rpa反应时间为10分钟(如图5所示)。将上述第一部分和第二部分联合,即可构建基于cas 12a酶和rpa技术联用的快速低丰度检测插入和缺失突变的方法,整个检测过程不超过2.5小时(如图6所示)、且灵敏度高(约为0.001%);克服了现有技术检测过程复杂且耗时长的缺点(现有的检测技术例如测序或者ddpcr,测序检测过程繁琐、灵敏度低、操作复杂且耗时长(检测耗时一周左右),ddpcr灵敏度高(约为0.001%),但其仪器昂贵,操作复杂,耗时长(检测耗时几天)。
76.参阅附图可知:一种快速低丰度检测插入和缺失突变的方法,包括如下步骤:
77.探针为单链dna,一端标记荧光基团,另一端标记猝灭基团,探针长度设计为6-8个核苷酸残基,te buffer稀释液稀释探针粉末,配置成液体,然后用nanodrop仪器测定浓度至20um,然后配置成5um的液体,待用;
78.本发明中的探针序列长度为6-8个碱基,没有碱基要求,具有通用性;
79.步骤二:制备cas12a(cpf1)核酸酶,浓度为2000pmol,使用超纯水稀释100倍,然后待用;
80.步骤三:挑选临床上最为常见的突变基因作为验证靶点,在pubmed网站上查询缺失突变点基的缺失突变点序列,
81.突变基因包括缺失突变基因、插入突变基因;缺失突变基因包括缺失突变egfr外显子19(-ggaattaagagaagc)、缺失突变cd41-42(-cttt);缺失突变点egfr外显子19为与肺癌相关的缺失突变,在其他疾病中的这个缺失突变也使用;
82.插入突变基因包括插入突变egfr20ins;任意缺失突变基因或插入突变基因均为
ggaattaagagaagc)缺失突变的原始序列3
‘
端和5
‘
端设计grna,分别在3
‘
端和5
‘
端设计9-10个碱基,即grna序列与突变链匹配的碱基数为18-20个碱基,总长度为40-42个碱基。
103.进一步地,在步骤三中,当突变基因选用缺失序列egfr外显子19(-ggaattaagagaagc)时,突变链的长度为106核苷酸残基,野生链的长度为121核苷酸残基,grna长度为40个核苷酸残基。
104.进一步地,当突变基因选用缺失序列egfr外显子19(-ggaattaagagaagc)时,
105.突变链的长度为106核苷酸残基,序列为:
106.gggactctggatcccagaaggtgagaaagttaaaattcccgtcgctatcaaaacatctccgaaagccaacaaggaaatcctcgatgtgagtttctgctttgctgtg,其互补序列为cacagcaaagcagaaactcacatcgaggatttccttgttggctttcggagatgttttgatagcgacgggaattttaactttctcaccttctgggatccagagtccc,
107.野生链的长度为121核苷酸残基,序列为:
108.gggactctggatcccagaaggtgagaaagttaaaattcccgtcgctatcaaggaattaagagaagcaacatctccgaaagccaacaaggaaatcctcgatgtgagtttctgctttgctgtg,其互补序列为cac agc aaa gca gaa act cac atc gag gat ttc ctt gtt ggc ttt cgg aga tgt tgc ttc tct taa ttc ctt gat agc gac ggg aat ttt aac ttt ctc acc ttc tgg gat cca gag tcc c,
109.grna长度为40个核苷酸残基为例,序列为:
110.uaauuucuacuaaguguagauggagauguuuugauagc ga。
111.进一步地,在步骤十二中,rpa反应的引物的设计原理为:
112.扩增长度在100-130bp之间,引物长30-35个碱基,不要出现聚嘌呤和嘧啶,且gc含量在30-70%之间,避免形成二级结构,发卡结构等。
113.进一步地,当突变基因选用缺失序列egfr外显子19(-ggaattaagagaagc)时,依据合成的egfr外显子19(-ggaattaagagaagc)基因突变链的长度为106和野生链的长度为121的核苷酸残基序列,设计rpa反应的引物,
114.引物序列为fp:gggactctggatcccagaaggtgagaaag,
115.rp:cacagcaaagcagaaactcacatcgaggatttc。
116.进一步地,在步骤五中,配制25ul的cas12a/grna混合反应液:2.8ul的2.1buffer,2.2ul的步骤二待用液,0.94ul grna(1um)和19.06的超纯水,待用。
117.进一步地,在步骤一中,探针的长度一般设置为6-8个核苷酸残基;探针的序列为ta碱基交错排列。
118.更进一步地,在步骤一中,探针长度设计为8核苷酸残基,探针的序列:ttattatt。
119.更进一步地,在步骤一中,荧光基团及其猝灭基团应当满足荧光基团的发射光谱与猝灭基团的吸收光谱有效重叠;
120.荧光基团及其猝灭基团至少选自fam与tamra,fam与bhq1,tet与bhq2。
121.实施例
122.实施例一
123.本实施例中,基于cas 12a酶和rpa技术联用的快速低丰度检测插入和缺失突变的方法,包括如下步骤:
124.步骤一:制备双标记荧光探针,
125.探针为单链dna,一端标记荧光基团,另一端标记猝灭基团,探针长度设计为8个核苷酸残基(序列为ta碱基交错排列),te buffer稀释液稀释探针粉末,配置成液体,然后用nanodrop仪器测定浓度至20um,然后配置成5um的液体,待用;
126.本实施例中的探针序列长度为8个碱基,没有碱基要求,具有通用性;本实施例中的,探针的序列:ttattatt;
127.步骤二:制备cas12a(cpf1)核酸酶,浓度为2000pmol,使用超纯水稀释100倍,然后待用;
128.步骤三:本实施例选用的突变基因为缺失突变点egfr外显子19(-ggaattaagagaagc)基因;
129.挑选临床上最为常见的一个缺失突变点egfr外显子19(-ggaattaagagaagc)基因,在pubmed网站上查询egfr外显子19(-ggaattaagagaagc)缺失突变点序列,
130.依据实验原理分别设计所需的突变链,野生链,grna;
131.突变链的长度为106核苷酸残基,序列为:
132.gggactctggatcccagaaggtgagaaagttaaaattcccgtcgctatcaaaacatctccgaaagccaacaaggaaatcctcgatgtgagtttctgctttgctgtg,其互补序列为cacagcaaagcagaaactcacatcgaggatttccttgttggctttcggagatgttttgatagcgacgggaattttaactttctcaccttctgggatccagagtccc,
133.野生链的长度为121核苷酸残基,序列为:
134.gggactctggatcccagaaggtgagaaagttaaaattcccgtcgctatcaaggaattaagagaagcaacatctccgaaagccaacaaggaaatcctcgatgtgagtttctgctttgctgtg,其互补序列为cac agc aaa gca gaa act cac atc gag gat ttc ctt gtt ggc ttt cgg aga tgt tgc ttc tct taa ttc ctt gat agc gac ggg aat ttt aac ttt ctc acc ttc tgg gat cca gag tcc c,
135.本实施例中grna长度为40个核苷酸残基,序列为:
136.uaauuucuacuaaguguagauggagauguuuugauagcga;
137.步骤四:用te buffer稀释液稀释步骤二所设计合成的链粉末,配置成液体,然后用nanodrop仪器测定浓度至20um,然后配置成2um的野生链和突变链的双链dna液体;
138.在配置成野生链和突变链的双链dna液体时:通过升温退火的方法制备成双链,升温85
°
,退火55
°
,置于37
°
下10分钟;
139.步骤五:配制cas12a/grna混合反应液;
140.配制25ul的cas12a/grna混合反应液:2.8ul的2.1buffer,2.2ul的步骤二待用液,0.94ul grna(1um)和19.06的超纯水,待用;
141.步骤六:将步骤四中的液体配制成双链的野生链和突变链,浓度为0.5um,待用;
142.配制成双链的野生链和突变链的方法为:通过升温退火的方法制备成双链,升温85℃,退火55℃,置于37℃下10分钟;
143.步骤七:配制50ul的待检反应体系:5ul的2.1buffer,5ul的步骤五待用液,2ul的步骤六待用液,2ul的mgso4(10mmol),2ul的步骤一待用液,34ul超纯水;
144.步骤八:将步骤七的待检反应体系放置于仪器中检测荧光曲线,读取数据,如图3所示;
145.步骤九:分别使用egfr外显子19(-ggaattaagagaagc)基因缺失突变的合成野生链
来稀释突变链,制备一系列浓度为0.5um混合样本,使其突变丰度范围为0.1%、0.01%、0.001%、0%;
146.步骤十:配制50ul的待检反应体系:5ul的2.1buffer,5ul的步骤五待用液,2ul的步骤九待用液,2ul的mgso4(10mmol),2ul的步骤一待用液,34ul超纯水;
147.步骤十一:将步骤十的待检反应体系放置于仪器中检测荧光曲线,读取数据,如图4所示;
148.步骤十二:依据合成的egfr外显子19(-ggaattaagagaagc)基因突变链的长度为106和野生链的长度为121的核苷酸残基序列,设计rpa反应的引物,
149.引物序列为fp:gggactctggatcccagaaggtgagaaag,
150.rp:cacagcaaagcagaaactcacatcgaggatttc;
151.步骤十三:用te buffer稀释液稀释步骤十二所设计合成的链粉末,配置成液体,然后用nanodrop仪器测定浓度至50um,然后配置成10um的液体;
152.步骤十四:配置rpa反应体系:分别向试剂盒的rpa反应管中加入稀释2000倍的步骤九的0.5um的0.1%、0.01%、0.001%、0%混合液体,2ul的rpa反应的引物fp和rp至于管盖,此步骤为冰上操作;然后盖上管盖,离心,离心管离心速度为1000转/min;然后37℃条件下反应10分钟;
153.步骤十五:配制50ul的待检反应体系:5ul的2.1buffer,5ul的步骤五待用液,20ul的步骤十四待用液,2ul的mgso4(10mmol),2ul的步骤一待用液,16ul超纯水;
154.步骤十六:将步骤十五的待检反应体系放置于仪器中检测荧光曲线,读取数据,如图6所示。
155.本实施例的检测灵敏度约为0.0011%,检测过程耗时2.0小时,检测成本约二十元钱。
156.实施例二
157.本实施例基于cas 12a酶和rpa技术联用的快速低丰度检测插入和缺失突变的方法同实施例一,不同之处在于:在步骤三中,grna序列:uaauuucuacuaaguguagau aaacauuacaaccccauaaa;
158.wt序列(即野生链序列):ccc tga ttt ggt caa tat gtg tac aca tat taa aac att aca ctt taa ccc ata aat atg tat aat gat tat gta tca att aaa aat aaa aga aaa taa agt agg gag att atg;
159.wt互补序列(即野生链互补序列):cat aat ctc cct act tta ttt tct ttt att ttt aat tga tac ata atc att ata cat att tat ggg tta aag tgt aat gtt tta ata tgt gta cac ata ttg acc aaa tca ggg;
160.mt序列(即突变链序列):ccc tga ttt ggt caa tat gtg tac aca tat taa aac att aca aa ccc ata aat atg tat aat gat tat gta tca att aaa aat aaa aga aaa taa agt agg gag att atg;
161.mt互补序列(即突变链互补序列):cat aat ctc cct act tta ttt tct ttt att ttt aat tga tac ata atc att ata cat att tat ggg tt tgt aat gtt tta ata tgt gta cac ata ttg acc aaa tca ggg。
162.本实施例的检测灵敏度约为0.0010%,检测过程耗时2小时,检测成本约二十元
钱。
163.实施例三
164.本实施例基于cas 12a酶和rpa技术联用的快速低丰度检测插入和缺失突变的方法同实施例一,不同之处在于:在步骤三中,grna序列:uaauuucuacuaaguguagau acaauaugugcuucuacaca;
165.wt序列:gcg aaa ggt tcg cgc ttg gaa gcg att gac ggt tgg caa act ttc taa ggt aca gga gac tgt gta gaa gag gcc aca tat tgt taa att gtc tag cta gtt ccc ggt aaa tca gc;
166.wt互补序列:gct gat tta ccg gga act agc tag aca att taa caa tat gtg gcc tct tct aca cag tct cct gta cct tag aaa gtt tgc caa ccg tca atc gct tcc aag cgc gaa cct ttc gc;
167.mt序列:gcg aaa ggt tcg cgc ttg gaa gcg att gac ggt tgg caa act ttc taa ggt aca gga gac tgt gta gaa gc aca tat tgt taa att gtc tag cta gtt ccc ggt aaa tca gc;
168.mt互补序列:gct gat tta ccg gga act agc tag aca att taa caa tat gtg ctt cta cac agt ctc ctg tac ctt aga aag ttt gcc aac cgt caa tcg ctt cca agc gcg aac ctt tcg c。
169.本实施例的检测灵敏度约为0.0012%,检测过程耗时2小时,检测成本约二十元钱。
170.实施例四:现有检测方法
171.通用型探针熔解曲线法目前是一种新型的基因突变检测技术,本实施例采用通用型探针熔解曲线法对缺失突变点egfr外显子19(-ggaattaagagaagc)基因进行插入和缺失突变检测。
172.本实施例采用通用型探针熔解曲线法的检测方法,具体步骤如下:
173.1)合成环状双标记荧光探针probe(序列为:
174.fam-cgcgcgggcccagccccttccgcgcgcg-bhq1)。
175.2)设计合成egfr外显子19(-ggaattaagagaagc)基因突变链位点基因的突变链-4,野生链-4,桥梁链-4(具体序列见下表1)。
176.3)使用te buffer将突变链-4,野生链-4,桥梁链-4溶解,配置成液体;
177.4)使用野生链-3稀释突变链-3,制备一系列浓度为100nm混合样本,使其突变丰度范围为100%至5%;
178.5)向4)中配置的混合样本中加入桥梁链-4,桥梁链-4的浓度为100nm,然后进行升温退火;
179.6)将5)中的混合液在37
°
下静置30min;
180.7)在6)的混合液中加入步骤一制得的探针,其最终浓度为100nm;
181.8)将7)的液体加入到检测管中,离心,然后将检测管放置于pcr仪器中检测熔解曲线。
182.本实施例中的条链序列见表1。
183.表1本实施例中的条链(即突变链-4,野生链-4,桥梁链-4)序列
[0184][0185][0186]
本实施例得到egfr外显子19(-ggaattaagagaagc)基因突变链的荧光曲线如图7所示。本实施例采用通用型探针熔解曲线法方法的检测限比较高,灵敏度不高、难以满足临床对低丰度突变检测的要求。
[0187]
通过实施例1-实施例4对比,可以看出,本发明方法在检测低丰度突变具有很大的优势。
[0188]
其它未说明的部分均属于现有技术。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1