一种阿朴酯关键中间体C10醛酯的制备方法与流程
一种阿朴酯关键中间体c10醛酯的制备方法
技术领域
1.本发明属于化学合成技术领域,具体涉及一种阿朴酯合成的关键中间体c10醛酯的合成方法。
背景技术:
2.阿朴酯作为类胡萝卜素化合物中的一种,属于脂溶性黄色素,其化学结构是c30共轭不饱和碳链化合物,在食品和饲料着色领域被广泛应用。在饲料领域,阿朴酯可以使肉鸡皮肤和蛋黄着色,具有抗氧化和增强机体免疫力的功效,此外阿朴酯作为维生素a的来源之一,其维生素a转化活性是β-胡萝卜素的25%。在食品营养领域,阿朴酯可以作为奶油、油脂、果酱及饮品的着色剂,同时对于人体机能具有一定的营养价值。
3.在自然界中阿朴酯天然存在于蛋黄和玉米中,但是由于生物提取方法工艺复杂,难以实现工业化生产,所以目前主要是通过化学合成手段来实现阿朴酯的量产。在阿朴酯合成路线中,可以采用c20+c10路线,该合成路线c20可以从维生素a的合成体系中分离出来,相对来说原料来源较为方便,但是c10醛酯合成路线较为复杂,原料多,收率较低,三废排放量大,工艺成本较高等特点导致c10醛酯合成难以工业化:
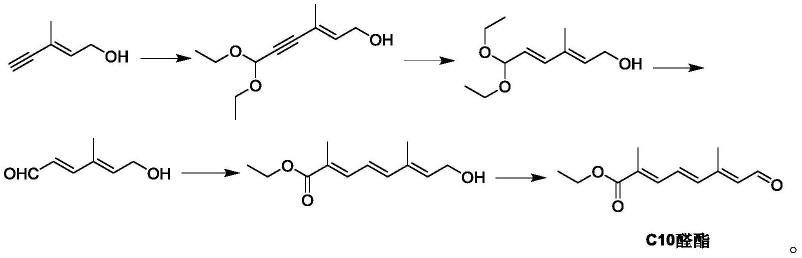
4.阿朴酯合成路线:c20+c10
[0005][0006]
已报道c10合成路线:
[0007][0008]
因此,有必要开发新的合成c10的方法。
技术实现要素:
[0009]
本发明的目的是提供一种阿朴酯合成过程中重要中间体c10醛酯的合成路线,该路线以乙酰乙醛、乙二醛、异丁酸乙酯为原料,经过选择性缩合、c10中间体合成、c10醛酯合成,经过三步反应即可得到c10醛酯,该路线简短,副产物少,合成成本低,收率优异。
[0010][0011]
为实现上述目的,本发明所采用技术方案如下:
[0012]
本发明第一方面提供了c6双醛中间体的合成,该反应以乙酰乙醛和乙二醛为原料,在强碱作用下发生选择性缩合反应得到c6双醛中间体;
[0013]
优选地,本发明所述c6双醛中间体的制备方法,包括以下步骤:
[0014]
1)在反应器中加入乙酰乙醛和反应溶剂,氮气置换多次,将反应液温度降至反应温度,加入强碱催化剂,然后开始滴加乙二醛溶液,待滴加完毕后保温反应,取样分析待反应液中乙酰乙醛转化率>99%,该步反应结束;
[0015]
2)反应结束后,反应液采用连续减压精馏方法进行分离,精馏塔塔顶可以得到纯度在90~98%的c6双醛产品。
[0016]
所述c6双醛中间体的制备过程中,步骤1)中,所述反应溶剂为乙腈、甲醇、乙醇、异丙醇、正丁醇中的一种或多种,优选正丁醇;
[0017]
所述反应温度为-10~10℃,优选-5~5℃;
[0018]
所述强碱催化剂为双(三甲基硅烷基)氨基钾和双(三甲基硅烷基)氨基钠中的一种或多种,优选双(三甲基硅烷基)氨基钾;
[0019]
所述乙二醛溶液(优选乙二醛的乙醚溶液)滴加时间为1~6h,优选2~3.5h;
[0020]
所述滴加完毕后保温反应的温度在10~28℃,优选15~20℃;
[0021]
所述乙酰乙醛和反应溶剂质量用量比为1:2~15,优选1:5~10;所述乙酰乙醛和乙二醛的质量用量比为1:0.70~2.5,优选1:0.9~1.5;所述乙酰乙醛与强碱催化剂质量用量比为1:0.5~1.5,优选1:0.9~1.2;
[0022]
步骤2)中,精馏塔的温度为45~85℃,优选55~70℃;压力为10~30kpa,优选15~20kpa;理论塔板数为10~30块,回流比为1~12,优选理论塔板数为12~18,优选回流比为5~9。
[0023]
本发明第二方面,涉及以异丁酸乙酯和c6双醛为原料制备c10醛酯,首先,异丁酸乙酯在溴代试剂三溴化吡啶作用下发生溴化,溴代物与镁粉反应得到格氏试剂,然后与c6双醛中间体反应得到c10中间体;然后脱除溶剂在过量碱试剂作用下c10中间体发生还原消
除反应,得到c10醛酯产品;
[0024]
优选地,包括以下步骤:
[0025]
1)在反应器中加入异丁酸乙酯、反应溶剂a预热至一定温度,然后加入三溴化吡啶进行预溴化,然后加入镁粉,进行氮气置换多次,反应一段时间后滴加c6双醛中间体,待其滴加完毕升温进行反应,取样进行分析,待异丁酸乙酯完全转化后结束反应;
[0026]
2)向步骤1)反应结束后的反应液中加入醋酸淬灭掉反应体系中的多余的镁粉以及有机镁化合物;然后向反应液中加入磷酸,升高温度继续进行反应得到c10中间体;
[0027]
3)将步骤2)中反应结束后的反应液减压蒸馏脱除反应溶剂a,加入反应溶剂b和过量碱试剂在室温下进行反应,取样分析,待反应液中无c10中间体时,结束反应。
[0028]
步骤1)中,所述反应溶剂a为乙醚或四氢呋喃中的一种或多种,优选四氢呋喃;
[0029]
所述预热温度为25~45℃,优选30~35℃;
[0030]
所述预溴化时间为1~5h,优选2~3.5h;
[0031]
加入镁粉后反应时间为0.5~3h,优选1~2h;
[0032]
所述c6双醛中间体滴加时间为1~6h,优选2~4h;滴加完毕后升温至40~80℃,优选50~75℃;
[0033]
所述异丁酸乙酯与反应溶剂a质量用量比为1:2~8,优选1:3~6;异丁酸乙酯与三溴化吡啶质量用量比为1:2.7~4.5,优选1:3.2~4.0;异丁酸乙酯与镁粉质量用量比为1:0.2~0.4,优选1:0.25~0.35;异丁酸乙酯与c6双醛中间体质量用量比为1:1~4,优选1:2~3;
[0034]
步骤2)中,初始镁粉与醋酸质量用量比为1:5~10,优选1:6~8;初始异丁酸乙酯与磷酸质量用量比为1:0.85~2.0,优选1:1~1.4;反应温度为85~120℃,优选90~100℃;
[0035]
步骤3)中,所述反应溶剂b为二氯甲烷、氯仿、dmso中的一种或多种,优选氯仿;初始异丁酸乙酯与反应溶剂b质量用量比为1:2~10,优选1:4~8;
[0036]
所述碱试剂为叔丁醇钠、叔丁醇钾、甲醇钠、甲醇钾中的一种或多种,优选叔丁醇钾;初始异丁酸乙酯与碱试剂质量用量比为1:2.5~5.0,优选1:3.0~4.0;
[0037]
所述碱试剂过量目的为中和步骤2)中的磷酸,同时为步骤3)提供碱催化剂。
[0038]
本发明的有益效果在于:
[0039]
1.采用价格低廉的原料乙酰乙醛和乙二醛,整个合成工艺成本低廉;
[0040]
2.该工艺涉及原料较少,路线简短,副产物少,且收率优异。
具体实施方式
[0041]
下面通过具体实施例对本发明的技术方案作进一步的说明,但并不局限于此。
[0042]
实施例1
[0043]
c6双醛的合成:
[0044]
在反应器中加入10g乙酰乙醛和80g正丁醇,氮气置换三次,将反应液温度降至-5℃,加入10g双(三甲基硅烷基)氨基钾,然后开始滴加乙二醛的乙醚溶液(40%浓度)30g,滴加时间为2h,待滴加完毕后在20℃下保温反应30min,取样分析待反应液中乙酰乙醛转化率>99%,该步反应结束;反应液采用连续减压精馏方法进行分离,控制精馏条件为:压力为15kpa、塔釜温度为65℃、塔板数为15块、回流比为9,塔顶可以得到纯度为93.6%的c6双醛
中间体,该步收率为85%。
[0045]
c10醛酯的合成:
[0046]
在反应器中加入10g异丁酸乙酯和50g四氢呋喃预热至30℃,然后加入31g三溴化吡啶预溴化3.5h,然后加入2.8g镁粉,进行氮气置换三次,反应2h后滴加12g c6双醛中间体,滴加时间为5h,待其滴加完毕升温至80℃进行反应,取样进行分析,待异丁酸乙酯完全转化后结束反应;向反应液中加入14.8g醋酸淬灭掉反应体系中的多余的镁粉以及有机镁化合物,然后加入9g磷酸,升温至95℃继续进行反应2h得到c10中间体;采用减压蒸馏脱除反应体系中的四氢呋喃和醋酸,加入50g氯仿和32g叔丁醇钾,在室温下反应,取样分析,待反应液中无c10中间体时,结束反应。经过气相分析该步收率为91.2%。核磁表征数据:1h nmr(300mhz,chloroform-d)δ1.16(m,3h),2.34(m,3h),2.42(m,3h),4.08(m,2h),6.01(s,1h),6.51(s,2h),7.22(s,1h),9.68(s,1h)ppm.
13
c nmr(101mhz,chloroform-d)δ191.1,167.2,154.8,142.8,134.4,130.1,128.7,127.4,61.7,14.2,13.0,11.8ppm.
[0047]
实施例2
[0048]
c6双醛的合成:
[0049]
在反应器中加入10g乙酰乙醛和50g乙腈,氮气置换三次,将反应液温度降至0℃,加入12g双(三甲基硅烷基)氨基钾,然后开始滴加乙二醛的乙醚溶液(40%浓度)52g,滴加时间为3h,待滴加完毕后在15℃下保温反应30min,取样分析待反应液中乙酰乙醛转化率>99%,该步反应结束;反应液采用连续减压精馏方法进行分离,控制精馏条件为:压力为15kpa、塔釜温度为80℃、塔板数为12块、回流比为6,塔顶可以得到纯度为91.8%的c6双醛中间体,该步收率为86.8%。
[0050]
c10醛酯的合成:
[0051]
在反应器中加入10g异丁酸乙酯和60g乙醚预热至25℃,然后加入32g三溴化吡啶预溴化2.5h,然后加入3.3g镁粉,进行氮气置换三次,反应2h后滴加30g c6双醛中间体,滴加时间为6h,待其滴加完毕升温至70℃进行反应,取样进行分析,待异丁酸乙酯完全转化后结束反应;向反应液中加入23.1g醋酸淬灭掉反应体系中的多余的镁粉以及有机镁化合物,然后向反应液中加入13.6g磷酸,升温至100℃继续进行反应2h得到c10中间体;采用减压蒸馏脱除反应体系中的乙醚和醋酸,加入50g二氯甲烷和39g甲醇钠,在室温下反应,取样分析,待反应液中无c10中间体时,结束反应。经过气相分析该步收率为94.3%。
[0052]
实施例3
[0053]
c6双醛的合成:
[0054]
在反应器中加入10g乙酰乙醛和100g甲醇,氮气置换三次,将反应液温度降至0℃,加入11g双(三甲基硅烷基)氨基钠,然后开始滴加乙二醛的乙醚溶液(40%浓度)50g,滴加时间为4h,待滴加完毕后在20℃下保温反应30min,取样分析待反应液中乙酰乙醛转化率>99%,该步反应结束;反应液采用连续减压精馏方法进行分离,控制精馏条件为:压力为12kpa、塔釜温度为50℃、塔板数为20块、回流比为10,塔顶可以得到纯度为92.0%的c6双醛中间体,该步收率为87.5%。
[0055]
c10醛酯的合成:
[0056]
在反应器中加入10g异丁酸乙酯和50g四氢呋喃预热至30℃,然后加入39g三溴化吡啶预溴化3h,然后加入2.4g镁粉,进行氮气置换三次,反应2h后滴加28g c6双醛中间体,
滴加时间为5h,待其滴加完毕升温至60℃进行反应,取样进行分析,待异丁酸乙酯完全转化后结束反应;向反应液中加入20.2g醋酸淬灭掉反应体系中的多余的镁粉以及有机镁化合物,然后向反应液中加入12.6g磷酸,升温至90℃继续进行反应2h得到c10中间体;采用减压蒸馏脱除反应体系中的四氢呋喃和醋酸,加入65g氯仿和35g叔丁醇钾,在室温下反应,取样分析,待反应液中无c10中间体时,结束反应。经过气相分析该步收率为94.1%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1