一种普乐沙福关键中间体的制备方法与流程
本发明属于药物合成,具体涉及一种普乐沙福关键中间的制备方法。
背景技术:
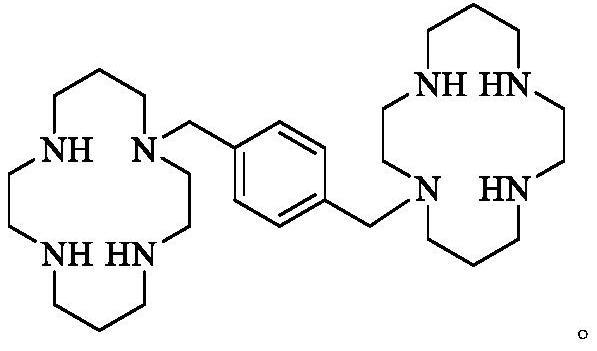
1、普乐沙福,英文名plerixafor,由美国健赞公司(genzyme corporation)开发,属于小分子单抗靶向药物。2008年12月普乐沙福被fda首次批准为罕用药物上市,临床研究表明,普乐沙福能大幅度提高患者的白细胞数量并促进造血干细胞从骨髓向血液流动,与粒细胞集落刺激因子(g-csf)有协同作用;已用于多发性骨髓瘤与非霍奇金淋巴瘤患者干细胞移植的临床治疗,其化学结构式如下:
2、
3、关于普乐沙福的制备公开的方法较多,大都以1,4,8,11-四氮杂环十四烷为原料,经过n1,n4,n8三保护,与1,4-二(卤代甲基)苯进行桥连,脱保护制得成品,合成路线如下:
4、
5、因此1,4,8,11-四氮杂环十四烷(cyclam)在多种合成策略中均被用作制备普乐沙福的关键中间体,cyclam可直接影响该药品的生产、目前关于cyclam的制备方法主要包括以下几种:
6、专利us5811544a中以1,3二氨基丙烷为原料,先和2equiv的氯乙酰氯反应生成酰胺,再接着和1equiv的1,3-二氨基丙烷反应环合,再用65%的红铝甲苯溶液还原得到cyclam。文献中反应步骤虽只有三步,但第二步1,3-二氨基丙烷和第一步生成的酰胺高温环合时间较长,需要24h,且环化产品不纯,产生如下所示的较多杂质,需要柱色谱纯化,限制了工业应用。合成路线如下:
7、
8、专利wo9705123a1中以二-(3-氨基丙基)乙二胺为原料,先和对甲苯磺酰氯发生磺酰化反应,再与1,2-二(对甲苯磺酰氧基)乙烷闭环,再用48%的氢溴酸和冰醋酸反应去除保护,最后碱化得cyclam。该方法优点是第一步取代及第二步环合收率较高,且第二步环合时间大大缩短,但脱ts其选用了48%氢溴酸/冰醋酸体系,收率只有72.0%,整体收率受到影响。合成路线如下:
9、
10、文献tetrahedron letters,1992,33(38):5505-5508中以二-(3-氨基丙基)乙二胺为原料,先和三氟甲磺酸酐发生磺酰化反应,再与1,2-二溴乙烷在碳酸钾作用下闭环,最后用钠/液氨脱除保护基,得到cyclam。该方法第一步用三氟甲磺酸酐作磺酰化试剂,收率不高;第二步所需环化温度110℃,温度较高易引起副反应发生;第三步脱三氟甲磺酰基,选用钠/液氨,且需在-33℃滴加,操作繁杂,不宜工业化;第二步和第三步合并总收率只有57.2%。合成路线如下:
11、
12、文献bulletin of the academy of sciences of the ussr division ofchemical science,36(2):372-376中以乙二胺为原料,和1,3-二溴丙烷发生取代反应生成二(2-氨基乙基)丙二胺,再在乙醇钠作用下和对甲苯磺酰氯发生磺酰化,再与1,3-二(对甲苯磺酰氧基)丙烷闭环,最后浓硫酸脱保护,经碱化得到cyclam。
13、但该方法以乙二胺为原料,和1,3-二溴丙烷发生取代反应生成二(2-氨基乙基)丙二胺,容易生成环戊烷副产物,该步收率不高,只有42.0%;在乙醇钠作用下和对甲苯磺酰氯发生磺酰化收率只有52.1%;与1,3-二(对甲苯磺酰氧基)丙烷环合收率65.3%;最后浓硫酸脱保护收率为81.0%。由上可见,该方法整体收率不高。合成路线如下:
14、
15、arkivoc,2006(4):212-233以1,3-丙二胺为原料,和乙二酸二乙酯环化成酰胺,再经硼烷还原,最后先酸化后碱化得到cyclam。该方法在用1,3-丙二胺和乙二酸二乙酯环化成酰胺后,用硼烷还原过程中使用超声,总收率只有19.0%,不宜工业放大。合成路线如下:
16、
17、鉴于目前制备cyclam时存在许多不足,因此,研究寻找一条操作简便、反应条件温和、操作过程安全、简便,产品收率高、纯度高的适合工业化生产cyclam的制备工艺仍是目前需要解决的问题。
技术实现思路
1、针对目前制备普乐沙福关键中间体1,4,8,11-四氮杂环十四烷时存在的诸多问题,本发明提供了一种新的1,4,8,11-四氮杂环十四烷的制备方法。该方法反应条件温和,操作过程安全、简便,所制得的目标产品具有较高的纯度、收率。
2、本发明的具体技术方案如下:
3、一种普乐沙福关键中间体1,4,8,11-四氮杂环十四烷的制备方法,其特征在于制备方法包括如下步骤:
4、步骤1:将浓硫酸滴加至化合物sm-1中,控温ta至反应结束后,加入甲苯回流带水至无水分蒸出后,将反应液降至室温,加入乙醇打浆,将所得固体减压真空干燥后即为化合物i-1;
5、步骤2:将化合物i-1加入碱的水溶液中,控温微沸反应,反应完毕后,将反应液中加入甲苯,90~95℃提取,合并有机相,饱和食盐水洗涤,有机相减压浓缩至干后即为化合物i-2。
6、步骤3:将化合物i-2、1,3-丙二胺加入醇水混合溶剂中,加入催化剂,控温tc反应,经检测反应完毕后,加碱调节ph至弱碱性,将反应液中加入甲苯,70~75℃提取,合并有机相,饱和碳酸钠溶液洗涤,饱和食盐水洗涤,有机相减压浓缩至干后即为目标产品i。。
7、合成路线如下:
8、
9、优选地,步骤1中所述的化合物sm-1与浓硫酸的投料摩尔比为1:2.2~2.8,优选1:2.4。
10、优选地,步骤1中所述的反应温度ta为40~80℃,优选45~50℃。
11、优选地,步骤2中所述的碱选自氢氧化钠、氢氧化钾中的一种,优选氢氧化钠。
12、优选地,步骤2中所述的化合物i-1与碱的投料摩尔比为1:6~14,优选1:9。
13、优选地,步骤3中所述的反应溶剂a选自叔丁醇-水、异丙醇-水、乙醇-水中的一种其组合,优选异丙醇-水。
14、优选地,步骤3中所述的催化剂选自甲酸、乙酸、三氟乙酸、苯磺酸中的一种。
15、优选地,步骤3中所述的化合物i-2、1,3-丙二胺、催化剂的投料摩尔比为1:1.3~2.0:0.8~1.2,优选1:1.5:1.0。
16、优选地,步骤3中所述的碱选自氢氧化钠、氢氧化钾、碳酸钠固体或其水溶液中的一种或其组合。
17、优选地,步骤3中所述的反应温度tc为70~80℃,优选75~80℃。
18、本发明的有益效果:
19、1.本发明提供了一条简便高效的制备普乐沙福关键中间体1,4,8,11-四氮杂环十四烷的方法,以n,n'-二(2-羟乙基)-1,3-丙二胺为起始物料,羟基先经硫酸活化,再在碱性条件下成环,再经1,3-丙二胺开环扩环环化后,制得目标产品。
20、2.该工艺通过改变反应条件有效避免ts-等保护基的引入及脱除,操作简便,缩短了反应步骤,减少了生产工时。
21、3.通过该工艺制得的目标产品具有较高的收率及纯度。
- 还没有人留言评论。精彩留言会获得点赞!