一种表达荧光基因的根瘤菌HH103及其构建方法与应用
一种表达荧光基因的根瘤菌hh103及其构建方法与应用
技术领域
1.本发明属于农用微生物的技术领域,具体涉及一种表达荧光基因的根瘤菌hh103及其构建方法与应用。
背景技术:
2.常规检测根瘤菌的侵染线只能通过光学显微镜在根毛发育早期通过显微镜在镜头下仔细的观察,需要很大的放大倍数,一根根根毛的仔细观察,需要很大的运气和耐心才能找到,侵染线,而通过荧光标记后在视野内是否有荧光的线出现则可以一目了然。
技术实现要素:
3.本发明的目的是为了更快更方便的找到根瘤菌的侵染线。
4.本发明提供一种表达荧光基因的根瘤菌hh103,所述表达荧光基因的根瘤菌hh103是以根瘤菌hh103为出发菌株,通过真核表达载体在出发菌株中表达荧光基因获得的。
5.进一步地限定,所述荧光基因为gfp基因、gus基因和rfp基因。
6.进一步地限定,所述gfp基因的序列如seq id no.3所示。
7.进一步地限定,真核表达载体为psoy10载体。
8.本发明提供一种上述的表达荧光基因的根瘤菌hh103的构建方法,所述构建方法的步骤如下:通过根瘤菌三亲杂交将含有gfp基因的回补荧光质粒转入根瘤菌hh103中获得的。
9.进一步地限定,所述含有gfp基因的回补荧光质粒的构建方法是:利用利用 atggtgagcaagggcgagga和cttgtacagctcgtccatgcc序列将gfp基因从 fu28载体上完成扩增,通过同源重组的方法将nptⅱ启动子与gfp基因克隆至pfaj1702上,所述nptⅱ启动子的基因序列如seq id no.4所示。
10.本发明提供一种检测根瘤菌侵染大豆能力的试剂盒,所述试剂盒含有上述的表达荧光基因的根瘤菌hh103。
11.本发明提供一种检测根瘤菌侵染线的形成的方法,所述方法的具体步骤如下:选取待检测的大豆毛状根材料,接种上述的表达荧光基因的根瘤菌hh103,通过荧光共聚焦显微镜观测并统计侵染线。
12.本发明提供上述的表达荧光基因的根瘤菌hh103在制备检测根瘤菌侵染大豆能力的试剂盒中的应用。
13.有益效果:常规检测根瘤菌的侵染线只能通过光学显微镜在根毛发育早期通过显微镜在镜头下仔细的观察,需要很大的放大倍数,一根根根毛的仔细观察,需要很大的运气和耐心才能找到,侵染线,而通过荧光标记后在视野内是否有荧光的线出现则可以一目了然。
附图说明
14.图1为gmmpk6:3
×
myc过表达载体构建;a:fu48 gmmpk6a:3
×
myc和fu48 gmmpk6b:3
×
myc的菌液pcr检测,泳道1为gmmpk6a,泳道2为gmmpk6b;m:trans 2k plus dna marker。b:psoy10 ubi:gmmpk6a:3
×
myc和psoy10 ubi:gmmpk6b:3
×
myc 的菌液pcr检测,泳道1为gmmpk6a,泳道2为gmmpk6b;m:trans 2k plus dna marker。
15.图2为gmmpk6s过表达毛状根qrt-pcr检测及免疫印迹分析;a:空载体、psoy10 ubi:gmmpk6a:3
×
myc和psoy10 ubi:gmmpk6b:3
×
myc毛根转化后,目的基因mpk6a和 mpk6b的相对表达量。使用gmukn1(glyma.12g020500)作为内参基因,数据为3次生物学重复的均值
±
标准误,student’s t test进行显著性分析,**代表p《0.01,***代表p《0.001。 b:免疫印迹分析阳性根,并使用actin抗体检测蛋白提取效率。ev代表psoy10空载、 oe-mpk6a代表psoy10 ubi:gmmpk6a:3
×
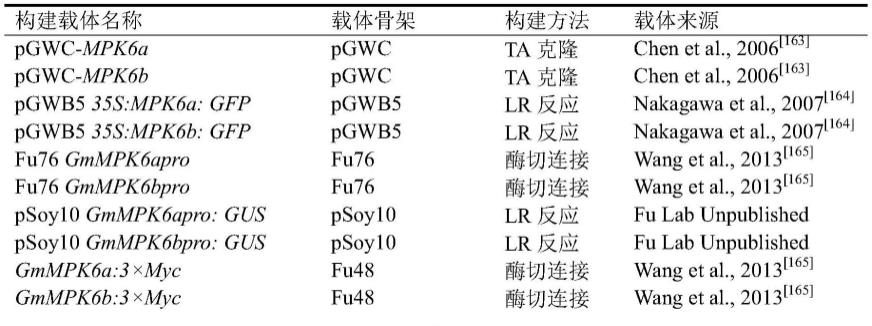
myc、oe-mpk6b代表psoy10 ubi:gmmpk6b:3
ꢀ×
myc。
16.图3为gmmpk6a和gmmpk6b过表达毛状根结瘤表型;a:psoy10 ubi:gmmpk6a:3
ꢀ×
myc、psoy10 ubi:gmmpk6b:3
×
myc和空载体分别接种hh103以及rhcn突变体的毛状根表型图,ev代表psoy10空载、oe-mpk6a代表psoy10 ubi:gmmpk6a:3
×
myc、oe-mpk6b 代表psoy10 ubi:gmmpk6b:3
×
myc,比例尺为1cm。b:毛状根转化并过表达gmmpk6 的根瘤表型的箱线图,使用student’s t test进行显著性分析,其中“***”代表p《0.001,“**”代表p《0.01。n=20。
17.图4为gmmpk6a和gmmpk6b基因编辑的结构示意;gmmpk6a和gmmpk6b基因编辑的结构示意图,包含sgrna的靶序列、pam位点信息以及mpk6a-cas-tf(r)和mpk6b-cas-tf(r)所在位置。
18.图5为gmmpk6a和gmmpk6b基因编辑情况检测;a:通过pcr鉴定检测5株基因编辑的大豆毛状根材料的编辑情况。m:dl5000 dnamarker;0:东农50野生型;1-15基因编辑的毛状根。b:经过测序后,确定的gmmpk6a和gmmpk6b基因编辑位点情况。
19.图6为敲除gmmpk6的结瘤表型;a:psoy10 gmmpk6-cas9和空载体psoy10分别接种hh103以及rhcn突变体毛状根表型图,ev:空载体psoy10;ko:knockout即psoy10 gmmpk6-cas9。比例尺为1cm。b:毛状根转化psoy10 gmmpk6-cas9的根瘤表型的箱线图,使用student’s t test进行显著性分析,其中“***”代表p《0.001,“*”代表p《0.05,“ns”代表p>0.05。n=15。
20.图7为gmmpk6表达模式,a是在根尖里表达;b是gmmpk6a与gmmpk6b在大豆根中的表达模式;
21.图8为pfaj1702 nptⅱ:gfp的pcr检测;pfaj1702 nptⅱ:gfp的菌液pcr检测m:trans 2k plusⅱdna marker。1:pfaj1702 nptⅱ:gfp。
22.图9为三亲杂交单克隆菌落的镜检;白色箭头所指的即是gfp阳性的单克隆菌落。
23.图10为gfp标记的根瘤菌的pcr检测;从上至下分别检测的基因为nptⅱ:gfp、ttsi 及其上游片段以及nifh,p:positive control,faj1702 nptⅱ:gfp;n:negative control, hh103野生型;m:trans 2k plus dnamarker。
24.图11为hh103和hh103-gfp的结瘤能力鉴定;a是hh103和hh103-gfp(28dpi) 的根瘤数统计。b:hh103和hh103-gfp(28dpi)的干重箱型图。c:hh103和hh103-gfp (28dpi)的根瘤豆血红蛋白含量柱状图。d:hh103和hh103-gfp(28dpi)的根瘤固氮酶活。a-d均使用t测验进行显著性分析,ns”代表p>0.05。
25.图12为过表达gmmpk6的侵染事件统计,a:构建的gfp标记根瘤菌菌液在荧光体式显微镜下的形态;b:foci、it以及rit的卡通模式图;c:荧光根瘤菌观测到的三种侵染事件示意图,比例尺均为20μm;d:过表达gmmpk6a和gmmpk6b的大豆毛状根在接种荧光标记根瘤菌后的侵染事件统计箱线图(24hpi)。foci代表侵染点;it代表未进入皮层的侵染线;rit代表已经进入皮层细胞的侵染线;ev代表psoy10空载;oe-mpk6a代表psoy10 ubi:gmmpk6a:3
×
myc;oe-mpk6b代表psoy10 ubi:gmmpk6b:3
×
myc;24hpi代表接种荧光标记根瘤菌24h;“***”代表p《0.001“ns”代表p>0.05。n=24。
具体实施方式
26.大豆根瘤菌hh103 rhcn突变体记载在函西,吕浩,郭广雨,等.大豆根瘤菌hh103 rhcn 突变对结瘤能力的影响[j].中国农业科学,2021,54(6):13.
[0027]
hh103ωttsi(效应因子不表达突变体)记载在田博宇,孙志君,刘函西,等.根瘤菌ttsi 突变体构建及结瘤表型鉴定[j].中国油料作物学报,2020,v.42;no.179(01):21-28.
[0028]
peasy-t1载体,helper(km)载体,pfaj1702(tet)载体和pgwc载体均为商业购买,商业购买。
[0029]
pjq200sk载体(gm)记载在quandt,j.,&hynes,m.f.(1993).versatile suicide vectorswhich allow direct selection for gene replacement in gram-negative bacteria.gene,127(1),15
–ꢀ
21.doi:10.1016/0378-1119(93)90611-6文献中。
[0030]
全基因组导入系群体(chromosome segments substitution lines,cssl)记载在文章_陈庆山,“作物回交导入系的构建与应用”。
[0031]
重组自交系群体(recombinant inbred lines,ril)记载在[1]王宏林.大豆重组自交系群体的构建,鉴定及其主要农艺性状qtl定位的研究[d].南京农业大学,2001.
[0032]
绥农14(suinong14,sn14),东农50(dongnong50,dn50)、野生豆zyd00006绥农 14、charleston、东农594、合00-23、红丰11、绥02-339、紫花2号、nattosan、黑农44、黑农35和北丰11均是黑龙江常见的品种,来自东北农业大学大豆生物学教育部重点实验室。
[0033]
快生型费氏中华根瘤菌hh103(源于西班牙塞维利亚大学francisco javier l
ó
pez-baena 实验室,记载在weidner s,becker a,bonilla i,et al.genome sequence of the soybeansymbiont sinorhizobium fredii hh103[j].journal of bacteriology,2012,194(6):1617.)。
[0034]
大肠杆菌(escherichia coli)菌株dh5α;根癌农杆菌(agrobacterium tumefaciens)菌株eha105;发根农杆菌(agrobacterium rhizogenes)菌株k599;快生费氏中华根瘤菌 (sinorhizobium fredii)菌株hh103,均是常见的菌种。
[0035]2×
ty液体培养基:胰蛋白胨(bacto-tryptone)16g、酵母提取物(bacto-yeast extract)10g、 nacl 5g、水1000ml,ph7.2,121℃灭菌30min.
[0036]
dpi,days post inoculation,接种后天数。
[0037]
hpi,接种后小时,hours post inoculation。
[0038]
fu系列载体包括fu48、fu76、fu76-cas9、fu79-gus以及fu79-2
×
sgrna均由傅永福研究员(中国农业科学院作物科学研究所)惠赠;植物表达载体psoy10由傅永福研究员(中国农业科学院作物科学研究所)惠赠;植物泛素启动子fu76-ubi由fu76改造,本实验室保存
提供。真核表达载体pgwb5由tsuyoshi nakagawa教授提供。
[0039]
载体骨架记载网站:tair-home page(arabidopsis.org)。
[0040][0041][0042]
fu79-2
×
sgrna是在fu79的载体上连接序列1:aagccaccatcatgcccatcgg和序列2: ccctccgtgaaatcaagctgctt。序列1和序列2是sgrna。序列1:aagccaccatcatgcccatcgg是 gmmpk6a的sgrna,序列2:ccctccgtgaaatcaagctgctt是gmmpk6b的sgrna。
[0043]
fu76-ubi载体是指在fu76载体上连接ubi序列。ubi序列如seq id no.7所示。
[0044]
ubifu48-融合3个myc标签是指在fu48载体上连接3个myc序列。3个myc序列如 seq id no.8所示。
[0045]
fu79-gus是指在fu79载体上连接gus序列。gus序列如seq id no.9所示。 fu76-cas9是指在fu76载体上连接gmmpk6a和gmmpk6b的sgrna序列。
[0046]
实施例1.构建mpk6基因过表达载体
[0047]
大豆材料均在25℃的长日照条件下(16hr光照/8hr黑暗)下恒温生长;本氏烟草在23℃的长日照条件恒温生长(16hr光照/8hr黑暗)。
[0048]
1.基因克隆及载体构建
[0049]
gmmpk6启动子载体构建:将gmmpk6转录起始位点(transcriptional start site,tss) 上游2000bp片段(gmmpk6apro,2000bp,seq id no.1)、gmmpk6b tss上游2000bp 片段(gmmpk6bpro,2000bp,seq id no.2),分别用ecor i和ecor
ⅴ
克隆位点酶切连接至入门载体fu76(lu m,cheng z,zhang x m,et al.spatial divergence of phr-pht1 modules maintains phosphorus homeostasis in soybean nodules[j].plant physiology,2020.),然后通过lr反应将目标片段与fu79-gus转移至真核表达载体psoy10上获得psoy10 gmmpk6apro:gus和psoy10 gmmpk6bpro:gus。
[0050]
gmmpk6过表达载体构建:将gmmpk6a的编码区(seq id no.10,glyma.02g138800) 和gmmpk6b的编码区(seq id no.11,glyma.07g206200)(coding sequence,cds),用mpk6a-f(r)和mpk6b-f(r)分别扩增,将gmmpk6a和gmmpk6b的cds通过ecor
ꢀⅴ
和kpn i克隆位点酶
切连接至入门载体fu48融合3个myc标签,然后通过lr反应将入门载体fu48上的gmmpk6a的编码区和gmmpk6b的编码区分别与fu76-ubi转移至真核表达载体psoy10上获得psoy10 ubi:gmmpk6a:3
×
myc和psoy10 ubi:gmmpk6b:3
×
myc。该部分载体用于大豆gmmpk6过表达的毛状根转化。
[0051]
为了验证gmmpk6对共生结瘤的影响,本研究通过构建植物过表达载体用以验证过表达gmmpk6调控大豆与根瘤菌共生体系建立。使用gmmpk6aoe-f(r)与gmmpk6aoe-f(r) 对目的片段进行克隆,电泳条带检测正确后,使用ecor
ⅴ
和kpnⅰ对fu48以及扩增片段进行酶切与连接并转化大肠杆菌,转化成功进行菌液pcr验证,见图(图1中的a)。随后通过gateway反应将fu48 gmmpk6a:3
×
myc和fu48 gmmpk6b:3
×
myc上的gmmpk6a的编码区和gmmpk6b的编码区分别与fu76-ubi的片段转移至双元载体psoy10,经菌液pcr 验证成功(图1中的b),测序成功后命名为psoy10 ubi:gmmpk6a:3
×
myc和psoy10 ubi:gmmpk6b:3
×
myc。
[0052]
2.毛状根转化:
[0053]
挑取饱满的大豆种子使用氯气熏蒸法灭菌8hr后取出置于超净台内吹净氯气,用灭菌的超纯水浸泡16hr至胚根伸长。
[0054]
将含有目标载体的k599农杆菌,挑取单克隆鉴定(转化及pcr鉴定方法同2.2.5.1), 活化后用yep培养基(5g
·
l-1
酵母提取物;10g
·
l-1
蛋白胨;5g
·
l-1
nacl;ph=7.0)振荡培养至菌液od
600
值在1.0左右,rcf=1700g离心20min,并重悬于lccm液体培养基(1/10
×
gamborg b5盐;30g
·
l-1
蔗糖;3.9g
·
l-1
mes;ph=5.4;灭菌后加入40mg
·
l-1
乙酰丁香酮);
[0055]
将浸泡好的大豆胚根部分切下,保留靠近子叶节的下胚轴和子叶,将外植体浸于重悬菌液中浸泡20min完成侵染。侵染后将外植体放于滤纸上吸干浸染液,而后转移至共培养基(1/10
×
gamborg b5盐;30g
·
l-1
蔗糖;3.9g
·
l-1
mes;4.25g
·
l-1
琼脂;ph=5.4;灭菌后加入400mg
·
l-1
cysteine;154.2mg
·
l-1
dithiothrietol;40mg
·
l-1
乙酰丁香酮),暗培养48hr。
[0056]
将共培养后的大豆外植体的下胚轴垂直插入含有毛状根诱导培养基(1
×
gamborg b5 盐;30g
·
l-1
蔗糖;0.59g
·
l-1
mes;7g
·
l-1
琼脂;ph=5.7;灭菌后加入100mg
·
l-1 timentin;100mg
·
l-1
cefotaxime)的组培瓶中,16hr光照、8hr黑暗下培养14d。
[0057]
待毛状根生长至2cm时,从培养基上移出,使用荧光体式显微镜筛选阳性根,剪去非阳性根,获得转基因毛状根用于后续实验。
[0058]
将psoy10 gmmpk6pro:gus系列表达载体;psoy10 ubi:gmmpk6:3
×
myc系列表达载体;导入发根农杆菌k599菌株中进行大豆毛状根转化,分别获得不同转基因的毛状根。
[0059]
3.过表达阳性毛状根的免疫印迹检测:提取镜检后的gmmpk6过表达阳性根液氮磨碎分装0.2g于1.5ml离心管中,加入500μl植物蛋白提取缓冲液(0.4m蔗糖、10mmtris-hcl ph=8.0、0.05%triton x-100以及1
×
蛋白酶抑制剂),冰浴30min后rcf=14000g 离心20min取上清。吸取100μl加入20μl的6
×
蛋白上样缓冲液(10%w/v sds、1mtris-hcl ph=8.0、30%v/v甘油、0.5m二硫苏糖醇以及10mm溴酚蓝)100℃煮沸解交联7min,短暂离心后进行sds-page垂直胶电泳,使用15%sds-page彩色凝胶配置试剂盒配置page凝胶,使用mops电泳缓冲液120v恒压30min。电泳结束后进行western blot 检测。
[0060]
为了进一步检测阳性根,利用qrt-pcr以及蛋白免疫印迹对阳性根进行检测,结果
见图(图2)分别过表达gmmpk6a和gmmpk6b后相应的基因具有显著表达。并且为了研究单一基因对结瘤表型的影响,同时通过分析同源基因的表达情况,即过表达gmmpk6a时分析内源gmmpk6b的表达,结果如图所示:同源基因没有显著差异表达。并且对镜检的阳性根进行免疫印迹检测使用肌动蛋白actin抗体作为内参抗体,检测蛋白提取效率,能看到清晰条带,结果表明psoy10 ubi:gmmpk6a:3
×
myc和psoy10 ubi:gmmpk6b:3
×
myc均已成功转化大豆毛状根,并对同源基因的表达没有显著影响,为单一表达,后续可以对单独基因进行根瘤表型的鉴定,用以研究基因gmmpk6a以及gmmpk6b过表达对结瘤表型的影响。
[0061]
实施例2.构建mpk6基因敲除载体
[0062]
1.gmmpk6的crispr-cas9载体构建:以gmmpk6a和gmmpk6b为靶基因,通过 crispr-p工具(http://crispr.hzau.edu.cn/cgi-bin/crispr2/crispr)设计靶基因的sgrna,每个靶基因挑选2个评分较高,距离较近,均在外显子区域的靶序列为crispr-cas9的编辑位点,然后合成两个靶序列片段序列1:aagccaccatcatgcccatcgg(seq id no.12),序列2: ccctccgtgaaatcaagctgctt(seq id no.13),(图5中的b)并分别用bsa i和bspq i连接到 fu79-2
×
sgrna载体,获得crispr-cas9的入门载体。最后通过lr反应将入门载体以及 fu76-cas9目标片段置换至psoy10上获得基因编辑载体psoy10 gmmpk6-cas9。
[0063]
通过过表达gmmpk6a和gmmpk6b的结果表明gmmpk6负调控共生结瘤,并且 gmmpk6a和gmmpk6b同源性较高,功能相似,为了进一步验证gmmpk6负调控共生结瘤。使用基因编辑技术进行gmmpk6a和gmmpkb的双敲除,通过crispr在线工具设计 2对靶基因的sgrna(序列1:aagccaccatcatgcccatcgg是gmmpk6a的sgrna,序列2: ccctccgtgaaatcaagctgctt是gmmpk6b的sgrna)分别用bsa i和bspq i连接至fu79 2
×
sgrna,测序成功后命名为fu79-sgrna-t1t2。之后通过gateway反应,将fu79-sgrna-t1t2以及 fu76-cas9的目的片段置换至psoy10内,测序成功命名为psoy10 gmmpk6-cas9,转入k599 菌株进行毛状根转化并进行基因编辑检测,用以评估编辑效率,基因编辑检测示意见图4。
[0064]
通过rfp荧光筛选阳性根,将5株基因编辑的大豆毛状根材料,每株选取3条阳性根与野生型东农50分别提取dna,利用mpk6cas1-f(r)(mpk6a),mpk6cas2-f(r)(mpk6b) 引物分别进行pcr扩增,进行基因编辑效率以及编辑方式检测,由于设计的两个靶点分别相距344bp和340bp,基因编辑时,两个靶点均被编辑时,有概率发生片段缺失产生见图(图 5中的a)。mpk6a与mpk6b均存在片段丢失,其中经过基因编辑有三分之一的mpk6a发生片段丢失。为进一步确定短片段为预期的基因编辑片段,通过连接t载体进行测序鉴定,结果如图(图5中的b)所示,符合实验预期。综上,获得的psoy10 gmmpk6-cas9载体可以发生预期的基因编辑,可以为后续的进一步mpk6基因功能研究提供材料基础,并有较好的编辑效率,可以用与毛状根转化验证敲除gmmpk6后的结瘤表型。
[0065]
2.毛状根转化:
[0066]
挑取饱满的大豆种子使用氯气熏蒸法灭菌8hr(小时)后取出置于超净台内吹净氯气,用灭菌的超纯水浸泡16hr至胚根伸长。
[0067]
将含有目标载体的k599农杆菌,挑取单克隆鉴定(转化及pcr鉴定方法同2.2.5.1),活化后用yep培养基(5g
·
l-1
酵母提取物;10g
·
l-1
蛋白胨;5g
·
l-1
nacl;ph=7.0)振荡培养至菌液od
600
值在1.0左右,rcf=1700g离心20min,并重悬于lccm液体培养基 (1/10
×
gamborg b5盐;30g
·
l-1
蔗糖;3.9g
·
l-1
mes;ph=5.4;灭菌后加入40mg
·
l-1
乙酰丁
香酮);
[0068]
将浸泡好的大豆胚根部分切下,保留靠近子叶节的下胚轴和子叶,将外植体浸于重悬菌液中浸泡20min完成侵染。侵染后将外植体放于滤纸上吸干浸染液,而后转移至共培养基 (1/10
×
gamborg b5盐;30g
·
l-1
蔗糖;3.9g
·
l-1
mes;4.25g
·
l-1
琼脂;ph=5.4;灭菌后加入400mg
·
l-1
cysteine;154.2mg
·
l-1
dithiothrietol;40mg
·
l-1
乙酰丁香酮),暗培养48hr。
[0069]
将共培养后的大豆外植体的下胚轴垂直插入含有毛状根诱导培养基(1
×
gamborg b5盐; 30g
·
l-1
蔗糖;0.59g
·
l-1
mes;7g
·
l-1
琼脂;ph=5.7;灭菌后加入100mg
·
l-1
timentin; 100mg
·
l-1
cefotaxime)的组培瓶中,16hr光照、8hr黑暗下培养14d。
[0070]
待毛状根生长至2cm时,从培养基上移出,使用荧光体式显微镜筛选阳性根,剪去非阳性根,获得转基因毛状根用于后续实验。
[0071]
psoy10 gmmpk6-cas9的大豆crispr cas9系列载体分别导入发根农杆菌k599菌株中进行大豆毛状根转化,分别获得不同转基因的毛状根。
[0072]
3.基因编辑效率检测:提取镜检后的gmmpk6基因编辑阳性毛状根液氮磨碎分装0.2g 于1.5ml离心管中,进行大豆dna提取。dna提取方法如下:
[0073]
(1)分装样品的1.5ml离心管加入小钢珠,震荡研磨仪60%额定功率研磨3min;
[0074]
(2)加入ctab提取液500μl,用涡旋振荡器摇匀,使组织粉末完全分散在ctab提取液中;
[0075]
(3)将1.5ml离心管置于烘箱中,65℃处理一个小时,期间轻摇数次;
[0076]
(4)上述反应液室温,rcf=12000g离心20min;
[0077]
(5)离心完成后,取上清液,加入600μl氯仿,离心机离心,rcf=12000g,离心20min;
[0078]
(6)取上清液,加入200μl的-20℃预冷异丙醇;
[0079]
(7)离心机离心rcf=12000g,离心20min弃上清,分别加入无水乙醇和75%乙醇洗涤 dna沉淀两次;
[0080]
(8)dna晾干后,加入灭菌水,待dna完全溶解后,-20℃保存。
[0081]
提取dna后,使用mpk6a-cas-tf(r)和mpk6b-cas-tf(r)引物进行pcr扩增,扩增产物经琼脂糖电泳检测后,产物回收ta克隆连接pgwc,进行测序验证,并与wm 82参考基因组比对。
[0082]
实施例3.根瘤菌荧光回补载体的构建
[0083]
1.使用gfp-f(r)将gfp从fu28载体上完成扩增,而后通过同源重组的方法将 nptⅱ启动子与gfp克隆至pfaj1702上,测序成功后,获得载体重新命名为pfaj1702 nptⅱ:gfp。或者将gus基因从fu28载体上完成扩增,而后通过同源重组的方法将nptⅱ启动子与gus克隆至pfaj1702上,测序成功后,获得载体重新命名为pfaj1702 nptⅱ:gus。或者将rfp基因从fu28载体上完成扩增,而后通过同源重组的方法将nptⅱ启动子与rfp克隆至pfaj1702上,测序成功后,获得载体重新命名为pfaj1702 nptⅱ:rfp。
[0084]
2.rna提取及qrt pcr
[0085]
大豆总rna提取:使用trizol对大豆组织的总rna提取,提取方法如下:
[0086]
(1)将研钵放在液氮预冷大豆组织取样材料放入研钵,在液氮中充分研磨并分装
于1.5mlrnase-freeep管中;
[0087]
(2)加入1ml的trizol液体于步骤(1)离心管中,冰上放置15min,漩涡混匀,室温静置10min;
[0088]
(3)4℃,rcf=12,000g,离心10min,吸取上清液于新的rnase-free离心管;
[0089]
(4)重复步骤(3),吸取200μl氯仿与离心管,漩涡混匀器混匀,冰浴10min;
[0090]
(5)4℃,rcf=12,000g,离心10min,弃上清液;
[0091]
(6)先后吸取700μl无水乙醇和300μldepc水到离心管中,4℃,rcf=12,000g,离心10min,弃上清并重复两次;
[0092]
(7)4℃短暂离心,吸尽上清,室温静置干燥;
[0093]
(8)加入50μldepc水,轻弹混匀;
[0094]
(9)60℃,2min水浴加速溶解,测定浓度;
[0095]
qrtpcr检测基因表达:对提取的大豆总rna进行cdna合成,使用hiscriptiireversetranscriptase反转录酶购自诺唯赞具体方法如下:
[0096]
(1)基因组dna的去除:在200μlrnase-freepcr管中加入下列成分:4
×
gdnawipermix,2μl;模板rna,1μl;rnasefreeddh2o,upto8μl,反应条件:42℃,2min;
[0097]
(2)rna反转录:在200μl离心管中加入下列成分:5
×
qrtsupermixii,2μl;步骤(1)反应液,8μl;反应条件:50℃,15min;85℃,2min;
[0098]
(3)得到的cdna-20℃保存用于后续检测基因表达。
[0099]
使用gmukn1(glyma.12g020500)做为定量内参基因,设计gmmpk6s及结瘤marker基因特异性定量引物,定量结果使用2-δδct
算法进行基因表达水平的相对定量分析,使用2-δct
算法进行gmmpk6组织特异性表达分析。
[0100]
在200μlrnase-freepcr管加入下列成分(20μl):chamquniversalsybrqpcrmastermix,10μl;上游引物(10μm),0.4μl;下游引物(10μm),0.4μl;cdna,100ng;rnasefreeh2o,upto10μl;real-timepcr反应采用二步法(2~3步设置35次循环)程序如下:95℃,20s;93℃,10s;65℃,20s。
[0101]
实施例4.荧光根瘤菌构建及鉴定
[0102]
gfp标记的hh103菌株构建:通过根瘤菌三亲杂交将回补荧光质粒pfaj1702nptⅱ:gfp转移至根瘤菌hh103中。根瘤菌三亲杂交具体步骤如下:
[0103]
将根瘤菌hh103和hh103ωrhcn划线与含rif(50mg/ml)的ty固体培养基,28℃培养直至长出单克隆;
[0104]
挑取单克隆于rif(50mg/ml)的ty液体培养基,活化根瘤菌,次日将回补载体菌株(pfaj1702nptⅱ:gfp,tet+)与自主转移菌株helper(prk2013,km+)划线与相应抗性的lb固体培养基上,过夜培养至长出单克隆;
[0105]
挑取回补菌株(pfaj1702nptⅱ:gfp,tet+)与自主转移菌株helper(prk2013,km+)单克隆于相应抗性的液体lb培养基,37℃活化;
[0106]
分别吸取50μl三种菌液与10ml相应培养基中培养od600值至0.6-0.8(根瘤菌od600值为0.3时,再培养两种大肠杆菌,三种菌od600值可几乎同时到0.6;
[0107]
三种菌培养到相同od600值,取三种菌各500μl于1.5ml离心管中,12000rpm离心1min,弃上清,1ml无抗ty重悬;
[0108]
调整根瘤菌:pfaj1702 nptⅱ:gfp:自主转移菌株的菌液比例为1:2:2,即重悬过后的 hh103菌液100μl,pfaj1702 nptⅱ:gfp菌株和自主转移菌株helper各200μl,加入到1.5 ml离心管中,12000rpm离心1min,弃上清,用30μl无抗ty重悬;
[0109]
将20μl的混合菌液滴到无抗ty固体培养基平板上28℃培养36hr-48hr;
[0110]
刮取菌苔到含有rif与tet的ty固体培养基28℃培养,直至长出单克隆;
[0111]
挑取单克隆于含抗生素的ty固体培养基28℃培养,进行筛选,重复2-3次。
[0112]
挑取筛选后的候选菌落于含有rif与tet的ty液体培养基中,28℃培养用于后续pcr 鉴定。
[0113]
gfp-tagged hh103的pcr鉴定:通过pcr鉴定候选菌液,鉴定无误后将gfp-tagged 的hh103命名为hh103-gfp。鉴定引物使用gfp-f(r)以及根瘤菌保守基因引物nifh-f(r) 和ttsi-f(r)。
[0114]
gus-tagged hh103的pcr鉴定:通过pcr鉴定候选菌液,鉴定无误后将gus-tagged的 hh103命名为hh103-gus。rfp-tagged hh103的pcr鉴定:通过pcr鉴定候选菌液,鉴定无误后将rfp-tagged的hh103命名为hh103-rfp。
[0115]
用到的引物序列如表1所示。
[0116]
1引物信息
[0117]
[0118][0119]
gfp基因序列(seq id no.3)。
[0120]
1.大豆结瘤试验:大豆于双层钵中培养至第一片三出复叶展开时(大豆毛状根移栽至双层钵后缓苗2-3天)分别接种快生根瘤菌(sinorhizobium fredii)hh103以及hh103ωrhcn,放置在温室中正常培养,培养条件大豆材料均在25℃的长日照条件下(16hr光照/8hr黑暗) 下恒温生长;本氏烟草在23℃的长日照条件恒温生长(16hr光照/8hr黑暗)。培养根瘤菌 hh103的ty培养基配方为:3g
·
l-1
酵母提取物,5g
·
l-1
蛋白胨,0.4g
·
l-1
氯化钙ph=7.0。植物低氮营养液配方:0.5μm硫酸锌、0.25μm硫酸镁、2μm硼酸、1mm氯化钙、1μm 硫酸锰、0.25μm硫酸钾、0.2μm硫酸铜、0.1μm硫酸钴、100μm磷酸二氢钾、10μm柠檬酸铁、0.1μm钼酸钠以及1μm硝酸钾,ph调至5.8。
[0121]
结果:过表达gmmpk6抑制结瘤发生:将携带有psoy10 ubi:gmmpk6a:3
×
myc和 psoy10 ubi:gmmpk6b:3
×
myc真核表达载体的发根农杆菌k599进行大豆毛状根转化,并使用psoy10空载作为阴性对照。由于psoy10载体存在dsred筛选标记,使用荧光体式显微镜镜检,去除非阳性根。
[0122]
筛选阳性根后分别接种根瘤菌hh103以及rhcn突变体,接种28d后统计根瘤表型,结果如图所示(图3),过表达gmmpk6a以及gmmpk6b在接种hh103和rhcn突变体后根瘤数以及根瘤干重显著减少。同时过表达gmmpk6a以及gmmpk6b在接种根瘤菌后瘤数上无显著差异,而瘤干重具有较大的差异,表明gmmpk6a和gmmpk6b在根瘤菌侵染期,具有相似功能,而gmmpk6b也作用于根瘤发育过程,结合启动子元件分析以及组织表达的 qrt-pcr,推测gmmpk6b可能在结瘤后期抑制根瘤的发育。综上,gmmpk6a与gmmpk6b 均负调控共生结瘤,并且在根瘤共生建立时行使相似的功能。
[0123]
接种rhcn突变体时,过表达gmmpk6a和gmmpk6b与ev相比,根瘤数明显减少,结合上文qrt-pcr结果表明,gmmpk6可能响应共生建立时的宿主免疫,而效应因子能够抑制gmmpk6表达。
[0124]
敲除gmmpk6促进共生结瘤:利用携带有psoy10 gmmpk6-cas9质粒的k599菌株,使用psoy10空载作为阴性对照,进行毛状根转化,进一步验证mpk6缺失后的结瘤表型。通过荧光体式显微镜,554nm激发光观测载体上荧光标签蛋白dsred,并去除非阳性根。分别接种hh103以及rhcn突变体,接种后28d统计根瘤数目以及根瘤干重结瘤表型,结果如图(图6)所
示。与过表达表型相反,敲除gmmpk6后,接种hh103根瘤数以及根瘤干重显著增加,进一步证明gmmpk6负调控大豆结瘤。而接种rhcn突变体时,敲除与空载体的毛状根根瘤数与根瘤干重均没有显著差异,结合转录组以及组织表达的结果,表明 gmmpk6可能通过介导效应因子的信号传递,并影响宿主免疫,从而影响大豆与根瘤菌共生的建立。
[0125]
2.gus染色:gus表达的组织化学染色主要参考jefferson等人的方法进行,并有所修改。将接种了s.fredii hh103与hh103ωrhcn的转基因发根植株生长48hr后,在流水下轻柔清洗地下部(保证至少10个独立的单株),获取的全根为新鲜组织为材料,将新鲜材料在-20℃预冷的90%丙酮中真空固定10min,在冰上静置20min后,用50mm磷酸缓冲液冲洗去除丙酮残留,用预先冷却的gus染液(50mm磷酸钠缓冲液,0.2%triton x-100,5mmk4fe(cn)6,5mm k3fe(cn)6,2mm x-gluc)冰上真空渗透10min,并在37℃下避光孵育12 hr。然后通过乙醇(15%,30%,60%,80%)梯度脱水,将样品在体式显微镜下观察。
[0126]
结果:为分析gmmpk6在大豆与根瘤菌共生建立时的组织表达模式,将含psoy10 gmmpk6apro:gus和psoy10 gmmpk6bpro:gus的转化毛状根植株移栽到蛭石生长2d后,分别接种hh103以及hh103ωrhcn两种菌株48h取其根进行gus染色,结果如图7中的 a所示gmmpk6在大豆根尖存在较高表达,同时gmmpk6a与gmmpk6b表达模式相似,在维管组织内存在更高表达,并且在接种根瘤菌hh103ωrhcn 48h后,转化gmmpk6apro 与gmmpk6bpro的gus积累量明显多于接种根瘤菌hh103的一组(图7中的b),表明接种hh103ωrhcn能够诱导gmmpk6a与gmmpk6b的表达,这部分结果与前文qrt-pcr分析结果相似,进一步证明rhcn的突变可以诱导mpk6的表达,并促进免疫反应。
[0127]
实施例5.荧光根瘤菌的应用
[0128]
根瘤菌侵染线观察:将psoy10 ubi:gmmpk6a:3
×
myc和psoy10 ubi:gmmpk6b:3
×
myc 的k599菌株,空载体psoy10作为对照,通过毛状根转化,获得gmmpk6a和gmmpk6b 过表达的毛状根材料,转移蛭石中培养2d后,接种gfp标记的根瘤菌hh103,每株选取3 条阳性根(24hpi),选择靠近下胚轴切口处1cm的根剪下,共8组重复。经组织透明化处理,组织透明化方法参考nadzieja等人clear see的方法,略有改进。获得的待检测组织置于4%多聚甲醛溶液中固定过夜,固定后使用清洗缓冲液(80mm hepes,ph=7.5)清洗3 次,每次10min,以便除尽甲醛。之后使用透明缓冲液(8m尿素、50%v/v甘油、0.5%tritonx-100以及40mm hepes ph=7.5)透明化处理,待组织完全透明后使用zeiss lsm700荧光共聚焦显微镜观察荧光,并统计侵染事件,包括侵染点,侵染线以及深入皮层的侵染线。
[0129]
过表达gmmpk6抑制侵染线的形成:大豆与根瘤菌共生建立离不开根瘤菌侵染线的形成,由于gmmpk6在侵染前期(12hpi)具有显著表达差异,并可能通过介导宿主免疫,抑制共生信号传导,推测gmmpk6会影响侵染线的形成,为了进一步分析gmmpk6对共生建立时的影响,本研究通过构建gfp标记的hh103用以观测统计gmmpk6对侵染事件影响。
[0130]
通过同源重组将细菌强启动子nptⅱ如seq id no.4所示,与从fu28上扩增的gfp克隆至pfaj1702上,并转化大肠杆菌,菌液pcr鉴定单克隆,条带大小均符合预期(图8),测序成功后,将获得载体命名为pfaj1702 nptⅱ:gfp。
[0131]
通过三亲杂交将pfaj1702 nptⅱ:gfp质粒转移至根瘤菌hh103中。简言之,将活化后的携带faj1702 nptⅱ:gfp质粒的大肠杆菌、helper菌株以及野生型根瘤菌按照2:2:1比例混合,在无抗ty固体培养基上28℃培养48h,刮取菌苔用500μl无抗ty培养基重悬,涂抹至
含有tet以及rif抗性的固体ty培养基上,28℃培养至长出单克隆,利用荧光体式显微镜筛选gfp阳性的单克隆,如图(图9)所示。挑取gfp为阳性单克隆进行菌液pcr 验证。由于野生型根瘤菌不含有nptⅱ:gfp片段,所以野生型并不能扩增该片段,另外,使用根瘤菌保守基因nifh以及ttsi基因上下游片段ttsi基因序列如seq id no.5,nifh基因序列seq id no.6分别扩增候选菌落,用以证明候选的阳性菌落为根瘤菌。结果如图10 所示:除16号以及17号菌液外,其余菌落的pcr产物大小均符合预期,表明完成pfaj1702 nptⅱ:gfp质粒的导入,结合荧光体式显微镜镜检结果,证明突变体构建成功。
[0132]
为确定荧光片段的导入对gfp标记hh103结瘤能力没有影响,对hh103以及 hh103-gfp进行结瘤能力鉴定,结果如图11所示:接种28天后,hh103与hh103-gfp在根瘤数、根瘤干重、豆血红蛋白含量以及固氮酶活性均没有显著差异,证明gfp荧光的导入不影响hh103的结瘤能力,可以用作后续侵染线观测的实验材料。
[0133]
使用上文提到的psoy10 ubi:gmmpk6a:3
×
myc和psoy10 ubi:gmmpk6b:3
×
myc的k599 菌株,空载体psoy10作为对照,通过毛状根转化,获得gmmpk6a和gmmpk6b过表达的毛状根材料,转移蛭石中培养2d后,接种gfp标记的根瘤菌hh103(24hpi)通过荧光共聚焦显微镜观测并统计侵染事件。即每株选取3条阳性根,选择靠近下胚轴切口处1cm的根剪下,共8组重复。经组织透明化处理后,通过荧光共聚焦显微镜,统计1cm根的全部侵染事件。侵染事件包括侵染点(foci,infection foci)、未进入皮层内部侵染线(its,infectionthreads)以及生长进入皮层内部的侵染线(rits,infection threads extending into a cortex cell),侵染事件模式图,以及观测到的实际的侵染事件见图12中的b和c,图12中的a是证明标记gfp的根瘤菌成功,并且可以发出绿色荧光。
[0134]
侵染事件统计结果如图(图12中的d)所示:过表达gmmpk6a和gmmpk6b后,侵染点以及未进入皮层的侵染线显著减少,表明过表达gmmpk6抑制侵染线的形成。ev (empty vector空载体)与过表达gmmpk6在生长进入皮层内部的侵染线(rits)表型上之间没有显著性差异,可能是由于观测时间仅为接种后24小时,生长进入皮层的侵染线较少,导致没有显著性差异。综上,结合上述结果可知,gmmpk6能响应根瘤菌侵染前期的免疫信号,通过影响侵染线的形成,抑制大豆结瘤。图12过表达gmmpk6的侵染事件统计。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1