一种用于COVID-19快速检测的双目标DNA步行器及其应用
一种用于covid-19快速检测的双目标dna步行器及其应用
技术领域
1.本发明属于分子生物学技术领域。更具体地,涉及一种用于covid-19快速检测的双目标dna步行器及其应用。
背景技术:
2.新型冠状病毒肺炎(coronavirus disease 2019,covid-19)是一种具有囊膜结构的正链rna病毒,具有传播性强、易变异及毒性强等特点。covid-19表面的刺突状蛋白(spike,s)即s蛋白,在病毒侵染宿主过程中扮演重要角色。s蛋白由s1亚基和s2亚基组成。其中s1亚基种含有受体结合域(receptor binding domain,rbd),该结构域可与人类受体蛋白血管紧张素转化酶2(angiotensin converting enzyme 2,ace2)结合,s2亚基介导病毒可与宿主细胞膜的融合。病毒在与受体结合后,可通过膜融合将遗传物质释放到宿主细胞中,从而完成对宿主的侵染。因此,s蛋白常作为重要的检测靶点用于抗原检测中。目前,covid-19已严重威胁全球公共卫生安全和健康,因此及时有效的早期快速检测对于筛查病毒患者、遏制传染及干预变异等具有重要意义。
3.病毒核酸阳性被作为sars-cov-2感染的确诊依据之一。目前,针对covid-19检测分为核酸检测法及抗原检测法。其中,荧光rt-pcr核酸检测法因具有较好的准确性是我国covid-19检测主流技术,但pcr过程需要使用精密仪器,成本高且耗费时间。相比之下,抗原检测法通过抗原抗体结合法,检测病毒外壳的特异性蛋白。该方法具有快速、操作简便的特点,不需要依赖专业机构,可实现居家自测并在30分钟内即可获得检测结果。但抗原检测法的灵敏度远不如荧光rt-pcr检测法,常会出现漏检或假阳性等问题。因此,能够克服检测条件限制、缩短检测时间、保证检测灵敏度及特异性,并降低假阳性干扰是covid-19检测技术研发的关键。
4.dna步行器是受启与自然界肌球蛋白、驱动蛋白和动力蛋白运动而构建的一种人工合成分子机器,由轨道链、步行链和稳定的驱动力三部分构成。在稳定驱动力下,步行者会沿预先设定好的轨道,无需人为干预,自发地进行渐进运动,以对特定的环境刺激作出响应。因此利用dna步行器可以在保证灵敏度及特异性识别的基础上,有效实现检测信号的放大,从而为目标物的快速灵敏检测搭建平台。目前,dna步行器还未被应用于covid-19检测中,为了避免covid-19检测中经常出现假阳性的问题,克服covid-19检测条件的限制、缩短检测时间、降低假阳性干扰,并及大地提高检测的灵敏度和特异性,因此,有必要研究和开发出更多更新的产品和方法,用于covid-19病毒核酸的快速检测,对于早期covid-19病毒筛查具有重要意义。
技术实现要素:
5.本发明为了提高covid-19检测的灵敏性、特异性和选择性,解决现有技术中covid-19快速检测过程中易出现假阳性的问题,提供一种用于covid-19快速检测的双目标dna步行器及其应用,极大缩短检测时间,且操作简易,无需复杂精密的仪器分析。
6.本发明的第一个目的是提供一种用于covid-19快速检测的双目标dna步行器。
7.本发明的第二个目的是提供所述双目标dna步行器的制备方法。
8.本发明的第三个目的是提供所述双目标dna步行器的应用。
9.本发明上述目的通过以下技术方案实现:
10.本发明提供一种用于covid-19快速检测的双目标dna步行器,所述双目标dna步行器包括:所述双目标dna步行器包括:表面可修饰dna的基材、行走链和若干信号链;所述行走链由捕获链1、捕获链2、游臂和阻滞链组成,捕获链1的5’端与基材表面连接,游臂的3’端连接阻滞链;信号链5’端与基材表面连接,信号链3’端修饰有生物素-辣根过氧化物酶标记物;所述捕获链1、捕获链2、游臂、阻滞链、信号链的序列依次如seq id no.1~5所示。
11.优选地,所述捕获链1的5’端及信号链5’端为巯基修饰。
12.优选地,所述表面可修饰dna的基材为由金纳米粒子、纳米磁珠或银纳米粒子覆盖的材料。
13.与现有技术中常规的dna步行器相比,本发明提供的双目标dna步行器中的信号链即为轨道链、本发明的行走链即为步行链,本发明由酶来提供稳定的驱动力。本发明基于银板分离技术,构建了一种可用于covid-19快速、灵敏检测的双目标dna步行器。本发明通过covid-19基因组设计引物,作为检测靶标:目标1(t1)和目标2(t2),并与其他的病毒或序列无高度相似,且具有极高的特异性和代表性。其中目标t1选自gene id:43740568第22828-22857位序列,目标t2选自gene id:43740568第21998-22027位序列,通过检测靶标t1和t2,来设计dna步行器的三个捕获探针捕获链1、捕获链2及游臂,进行特异性识别,以酶剪切提供稳定动力,使步行链自主运行。
14.本发明提供双目标dna步行器的制备方法,包含以下步骤:
15.s1.制备表面可修饰dna的基材;
16.s2.修饰基材:将步骤s1制备的基材浸泡在由捕获链1和信号链配制的溶液中,孵育,修饰连接后冲洗;
17.s3.将步骤s2已修饰的基材浸泡在含有链霉亲和素的溶液中,孵育、冲洗;再将基材浸泡到含有生物素-辣根过氧化物酶的溶液中,孵育、冲洗;
18.s4.将游臂和过量阻滞链混匀得游臂-阻滞链溶液,制备捕获链2溶液,即得用于covid-19快速检测的双目标dna步行器。
19.特别说明,在步骤s3中将基材浸泡在含有链霉亲和素溶液中是为了将链霉亲和素与基材上信号链的3’端生物素相连接;再将基材浸泡到含有生物素-辣根过氧化物酶的溶液中,与基材的信号链3’端连接形成生物素-链酶亲和素-生物素-辣根过氧化物酶。
20.优选地,所述基材为银纳米粒子覆盖的玻璃片,采用传统的银镜反应制得银纳米粒子覆盖的玻璃片(银板)。
21.优先地,步骤s2中捕获链1和信号链的摩尔比为1:10~30。
22.更优选地,捕获链1和信号链的摩尔比为1:20。
23.优选地,步骤s4中游臂-阻滞溶液中游臂和阻滞链摩尔比为1:1~1.5。
24.优选地,步骤s2中采用的生物素-辣根过氧化物酶的溶液浓度为120nmol/l。
25.本发明提供双目标dna步行器在制备covid-19快速检测产品中的应用。
26.本发明提供一种covid-19快速检测产品,含有上述双目标dna步行器。
27.优选地,所述covid-19快速检测产品含有限制性内切酶、h2o2和abts
2-。
28.更优选地,所述限制性内切酶为nb.bbvci酶。
29.优选地,abts
2-浓度为100~150nmol/l,体系反应时长为45~75min。
30.本发明提供的双目标dna步行器的信号链和捕获链1通过ag-s键自组装在银板(scg)表面,信号链的3’端连有链酶亲和素-生物素-辣根过氧化物酶(sa-biotin-hrp),与游臂(sw)部分互补,且存在核酸内切酶nb.bbvci的切割位点。当目标t1和t2同时存在时,捕获链1(cp1)和捕获链2(cp2)首先捕获t1,进而cp2和sw捕获t2,此特异性识别过程完成了cp1/cp2/sw步行链的组装,表明步行器已被t1、t2激活;进而促使步行链与信号链(sd)结合形成双链结构,通过加入的核酸内切酶nb.bbvci可识别切割双链结构中的切割位点,并释放hrp。该剪切过程释放的hrp可催化加入的h2o2与abts
2-之间的氧化还原反应,使abts
2-氧化为在416nm处存在紫外检测信号的abts
+
。因此该剪切过程亦是信号放大的过程,且除紫外检测信号外,会伴随出现可视化的颜色反应,最终将目标转化为紫外吸光度及实时颜色变化两种检测信号输出。
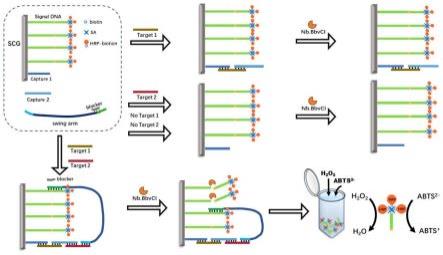
31.当仅存在t1时,cp1和cp2捕获t1,而sw处于游离状态,步行链组装失败,同时sw携带的阻滞链(bl)防止了其与sd的非特异性结合,因此hrp无法脱离银板,加入abts
2-无检测信号出现。当仅存在目标2、目标2或目标1、2均不存在时,则无法激活dna步行器运行,无检测信号出现。本发明通过巧妙地设计,实现了仅在双目标同时出现时才可激活步行器从而产生检测信号,极大提升了检测灵敏度和特异性。
32.本发明具有以下有益效果:
33.本发明提供了一种可用于covid-19快速检测的双目标dna步行器及其应用。本发明提供的双目标dna步行器通过行走链的三个捕获探针对检测目标1和目标2进行特异性识别,逐步完成行走链的组装,激活步行器,由酶剪切提供稳定动力,释放信号链携带的hrp,催化h2o2与abts
2-发生氧化还原反应,产生检测信号,能根据可视的颜色变化直接判断结果,极大的缩短检测时间、简化检出流程。本发明提供的双目标dna步行器在5
×
10-9-3.0
×
10-7
mol/l范围内呈良好的线性关系,线性方程为:y=0.0277+0.00252,检出限为1.19nmol/l,r2为0.995。
34.与传统核酸检测法相比,本发明筛选出只对covid-19病毒特异性识别的s蛋白的两段特异性的片段基因来设计捕获探针进行特异性识别,降低检测过程中地假阳性干扰,防止对单一的目标检测出现的假阴性或者是假阳性检测结果,并提高双目标dna步行器的特异性和灵敏性;同时,只有双目标同时出现才可激活步行器从而产生检测信号,极大提升了检测的特异性和选择性。并且本发明还设置有阻滞链,能够防止出现非目标激活的非特异性结合及剪切,进一步保证了本发明对t1、t2双目标同时的特异性识别,提高识别的灵敏度,减少假阳性结果。
35.本发明提供的双目标dna步行器具有优异的检测灵敏度、选择性,和易操作、耗时短等特点,为实现covid-19核酸的快速检测提供了新途径,对于早期covid-19病毒筛查具有重要意义,为covid-19家庭快检试剂盒的研发提供有效参考,同时也为快速、简易、超灵敏的生物传感检测平台的构建开拓新思路。
附图说明
36.图1为双目标dna步行器的cp1中隔离序列的碱基数目的优化;
37.图2为双目标dna步行器的sw中隔离序列的碱基数目的优化;
38.图3为双目标dna步行器的bl碱基数目的优化;
39.图4为双目标dna步行器的sd中隔离序列的碱基数目的优化;
40.图5为双目标dna步行器的dna序列反应流程图;
41.图6为双目标dna步行器的基本原理图;
42.图7为双目标dna步行器的可行性分析(a:0nmol/l t1+0nmol/l t2;b:300nmol/l t1+0nmol/l t2;c:0nmol/l t1+300nmol/l t2;d:300nmol/l t1+300nmol/l t2);
43.图8为双目标dna步行器的灵敏度分析;
44.图9为双目标dna步行器的特异性分析;
45.图10为捕获链1与信号链的比例优化;
46.图11为双目标dna步行器的反应时间优化;
47.图12为abts
2-浓度优化。
具体实施方式
48.以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
49.除非特别说明,以下实施例所用试剂和材料均为市购。
50.实际样本共10份咽拭子,采集自有临床症状的covid-19感染患者及其密切接触者,所有标本采集及运输严格遵从《新型冠状病毒实验室检测技术指南(第五版)》要求。
51.以下实施例采用缓冲溶液是pbs缓冲溶液(ph 7.0,10mmol/l,含100mmol/l nano3)和naac/hac缓冲溶液(ph 5.0,10mmol/l),所用水为超纯水,所有试剂均为分析纯并通过正规渠道购买。
52.本发明包含:目标1(target1,t1)、目标2(target2,t2)、信号链(signal dna,sd)、生物素-辣根过氧化物酶(biotin-hrp)、捕获链1(capture 1,cp1)、捕获链2(capture 2,cp2)、游臂(swimming arm,sw)、阻滞链(blocker,bl)、巯基(sh)、生物素(biotin)、链霉亲和素(sa)、游臂-阻滞链(swimming arm-blocker)、银板(scg)、生物素-辣根过氧化物酶(biotin-hrp)、链酶亲和素-生物素-辣根过氧化物酶(sa-biotin-hrp)。
53.本发明设计的双目标dna步行器序列如下表所示:
54.表1双目标dna步行器序列表
[0055][0056]
注:sh为巯基;biotin为生物素;
▲
为酶切位点;下划线为互补部分。
[0057]
实施例1双目标dna步行器的设计
[0058]
本发明基于银板分离技术,构建了一种可用于covid-19快速、灵敏检测的双目标dna步行器。本发明采用covid-19病毒s蛋白的两段特异性的片段基因作为检测靶标target 1和target2,来设计dna步行器的三个捕获探针capture 1、capture 2及swing arm,进行特异性识别,具体的设计方法如下所示。
[0059]
1、检测靶标的设计
[0060]
本发明的检测靶标来自covid-19基因组,且与其他的病毒或序列无高度相似,具有极高的特异性和代表性,作为检测靶标。其中目标t1选自gene id:43740568第22828-22857位序列,t1:ttataaattaccagatgattttacaggc tg;目标t2选自gene id:43740568第21998-22027位序列,t2:cacaaaaacaacaaaagttggatggaaagt。以此靶标序列进一步设计捕获链和游臂。
[0061]
2、捕获链的设计
[0062]
(1)捕获链1的序列设计
[0063]
捕获链的设计分为三个部分,第一部分为巯基修饰(-sh),cp1通过利用巯基与金属银形成的共价键,将带有巯基修饰(-sh)的cp1修饰至银板上。第二部分为隔离序列,将寡核苷酸链修饰至银板上后,会产生空间位阻效应影响杂交效率,从而对实验方法效率产生影响,因此,需设计隔离序列以减小空间位阻效应,以获得最优结合效率,促进实验方法构
建;同时,为降低背景值,避免非特异性杂交,依据目标t1、t2序列,对隔离序列设置新的序列组合,将隔离序列由传统poly t(或poly a)更改为aca cac重复出现的序列。
[0064]
第三部分为t1的捕获序列,捕获链cp1上还设计有对目标t1的捕获序列。当参与杂交的碱基数目大于12时,可保证杂交的稳定性,因此选取14个碱基构成cp1的t1捕获序列,即可保证cp1与t1之间的稳定杂交结合。基于碱基互补配对原则,此部分序列可与t1(5
’‑
ttataaattaccagatgattttacaggctg-3’)下划线部分特异性结合,实现对t1的特异性捕获。
[0065]
为减小空间位阻效应,以获得最优结合效率,对cp1中的隔离序列中的碱基数目进行优化,设置碱基数目分别为4、6、8、10、12的隔离序列在构建方法中的检测表现情况,结果如图1所示,可知在捕获链cp1上的隔离序列最优碱基数目为10。
[0066]
具体cp1序列设计如下所示:
[0067][0068]
其中,
①
巯基修饰(-sh),
②
隔离序列(碱基数目:10个;序列组成:5
’‑
aca cac aca c-3’),
③
t1捕获序列(碱基数目:14个;序列组成:5
’‑
cag cct gta aaa tc-3’)。
[0069]
(2)捕获链2的序列设计
[0070]
捕获链2的第一部分为目标t1的捕获序列,用于特异性捕获t1,同时完成cp2的自组装。cp2选取13个碱基构成t1捕获序列,即可保证cp2与t1之间的稳定杂交结合。同时基于碱基互补配对原则,此部分序列可与t1(5
’‑
ttataaattacca gat gattttacaggctg-3’)下划线部分特异性结合,实现对t1的特异性捕获。
[0071]
第二部分为设置隔离序列,用于进一步减小dna链在逐步杂交结合过程中存在的较小的空间位阻效应,降低背景值,获得最优结合效率。
[0072]
第三部分为t2捕获序列,用于特异性捕获t2,同时为sw的结合提供条件。选取13个碱基构成cp2的t2捕获序列,即可保证cp2与t2之间的稳定杂交结合,基于碱基互补配对原则,序列可与t2(5
’‑
cacaaaaacaacaaaag ttggatggaaagt-3’)下划线部分特异性结合,实现对t1的特异性捕获。
[0073]
具体cp2序列设计如下所示:
[0074][0075]
其中,
①
t1的捕获序列(碱基数目:13个;序列组成:5
’‑
tgg taa ttt ata a-3’),
②
隔离序列(碱基数目:3个;序列组成:5
’‑
cgc-3’),
③
t2捕获序列(碱基数目:13个;序列组成:5
’‑
act ttc cat cca a-3’)。
[0076]
3、游臂的设计
[0077]
游臂sw的设计分为三个部分,第一部分为t2捕获序列,用于特异性捕获t2,同时完成游臂sw的自组装。选取14个碱基构成sw的t2捕获序列,即可保证sw与t2之间的稳定杂交结合,基于碱基互补配对原则,序列可与t2(5
’‑
cacaaaaacaacaa aagttggatggaaagt-3’)下划线部分特异性结合,实现对t2的特异性捕获。
[0078]
第二部分为隔离序列,用于延长sw的长度,保证sw能够与信号链sd充分杂交。由于游臂长度主要依赖于隔离序列长度,游臂过短无法与sd充分结合,游臂过长影响反应时间,其长度能够完全切割已修饰的sd最为适宜。因此,对sw的碱基数目进行了优化实验,试验碱
基数目分别为25、30、33、35、40的隔离序列,在构建方法中的检测表现情况,结果如图2所示,在sw中隔离序列的最优碱基数目为33。同时,为降低背景值,避免非特异性杂交,依据目标t1、t2序列,对隔离序列设置新的碱基组合,将隔离序列由传统poly t或poly a,更改为aca cac重复出现的序列。
[0079]
第三部分为酶识别序列,与sd信号链特异性结合形成双链结构,使识别位点能够被nb.bbvci酶识别,满足酶剪切条件。由于nb.bbvci酶固定识别双链序列环境为(5
’‑
cctcagc-3’)和三角标注为酶切割位点,故设互补碱基数目为7,序列为cctcagc。
[0080]
具体sw序列设计如下所示:
[0081][0082]
其中,
①
t2的捕获序列(碱基数目:14个;序列组成:5
’‑
ttg ttg ttt ttg tg-3’),
②
隔离序列(碱基数目:33个;序列组成:cac aca cac aca cac aca cac aca cac aca cgc),
③
酶识别序列(碱基数目:7个;序列组成:5
’‑
cct cag c-3’)。
[0083]
4、阻滞链的设计
[0084]
阻滞链序列与sw(5
’‑
ttgttgtttttgtgcacacacacacacacacacacacacacacacgccctcagc-3’)下划线部分互补,保护酶切割识别位点,降低背景值,防止非目标引导下的sw与sd的非特异性结合。阻滞链bl过短无法实现保护作用,过长则会导致bl无法被sd置换,因此,为获得适宜的阻滞链碱基数目,进行了优化实验,设置碱基数目分别为7、8、9、10、11、12,在构建方法中的检测表现情况,结果如图3所示,可知阻滞链的最优碱基数目为10。所以,阻滞链的序列设计为:碱基数目:10个;序列组成:5
’‑
gag ggc gtg t-3’。
[0085]
5、信号链的设计
[0086]
信号链sd的设计分为五个部分,第一部分为巯基修饰(-sh),通过利用巯基与金属银形成的共价键,将带有巯基修饰(-sh)的sd修饰至银板上,组成该步行器的信号链。
[0087]
第二部分为隔离序列,将寡核苷酸链修饰至银板上后,会产生空间位阻效应影响杂交效率,从而对实验方法效率产生影响。因此,需设计隔离序列以减小空间位阻效应,以获得最优结合效率,促进实验方法构建;同时,为降低背景值,避免非特异性杂交,依据目标t1、t2序列,对隔离序列设置新的碱基组合,将隔离序列由传统poly t或poly a,更改为acacac重复出现的序列。为优化sd中的隔离序列中的碱基数目,试验碱基数目分别为12、13、16、17、18的隔离序列,在构建方法中的检测表现情况,结果如图4所示,可知隔离序列的最优碱基数目为16。
[0088]
第三部分为酶切割序列,用于特异性捕获sw,形成双链结构,使切割位点能够被识别,为后续酶剪切提供条件。根据nb.bbvci酶设置互补碱基数目为7,基于碱基互补配对原则,此部分序列可与sw序列(5
’‑
ttgttgtttttgtgcacacacacacacacacacacacacacacacgc cctcagc-3’)下划线部分特异性结合,从而使切割位点被识别。
[0089]
第四部分为隔离序列,减小因biotin修饰带来的较小的空间位阻效应,以促进实验方法构建。
[0090]
第五部分为生物素修饰(-biotin),通过修饰biotin连接辣根过氧化物酶(hrp),进而实现biotin-hrp的修饰,为后续检测信号修饰做铺垫。
[0091]
具体sd序列设计如下所示:
[0092][0093]
其中,
①
巯基修饰(-sh),
②
隔离序列(碱基数目:16个;序列组成:5
’‑
aca cac aca cac aca c-3’),
③
酶切割序列(碱基数目:7个;序列组成:),
④
隔离序列(碱基数目:2个;序列组成:5
’‑
aa-3’),
⑤
生物素修饰(-biotin)。
[0094]
结合上述的设计,本发明采用的双目标dna步行器的dna序列设计如图5所示,其中,cp1和cp2中的褐色部分序列与t1互补,因此t1的出现可被cp1与cp2捕获,从而实现cp1与cp2的自组装;cp2和sw中的绿色部分与t2互补,基于t1的捕获,t2的出现可被sw捕获,进而实现cp1/cp2/sw步行链自组装的完成,至此表明t1和t2的同时出现激活了步行器。sw中蓝色部分序列与sd的蓝色部分序列互补,sd序列中三角脚标为限制性内切酶剪切位点;因此,cp1/cp2/sw步行链可与sd相结合形成双链结构,并可在加入nb.bbvci后被切割,释放hrp;切割后步行链会结合新的sd,并再次切割,此过程由酶剪切提供稳定动力,自发进行直至sd被全部切割;因此该步骤亦为检测信号放大过程。阻滞链bl与sw划线部分序列结合,防止出现非t1、t2目标激活的非特异性结合及剪切,保证对t1、t2双目标同时的特异性识别,提高识别的灵敏度,减少假阳性结果。
[0095]
此外,本发明设计的cp1、sw、sd中的隔离序列,并未选择传统polyt作为隔离序列,为避免非特异性结合,依据目标t1、t2序列对隔离序列设置新的碱基组合。其次,以aca cac组合重复出现可完全与乱序的目标链的互补序列相区别开,避免与目标序列互补序列的情况出现,并同时满足不同的隔离长度需求,从而进一步提高其检测特异性,降低检测过程的假阳性。
[0096]
实施例2双目标dna步行器的制备
[0097]
1、银板的制备:
[0098]
采用传统的银镜反应制得银纳米粒子覆盖的玻璃片(银板)。石英玻璃片以及所有使用的玻璃仪器均需氢氧化钠溶液,硝酸以及乙醇彻底清洗,然后用超纯水冲洗干净并吹干。
[0099]
首先制备银氨溶液,在洁净的烧杯中加入5ml的3%agno3溶液,然后加入1%nh3·
h2o,滴加的同时振荡,直至混合液澄清。然后将1cm
×
1cm的玻璃板放置于银氨溶液和10%的葡萄糖混合溶液当中(银氨溶液体积:葡萄糖溶液体积=1:2),反应在25℃下反应10分钟;最后将反应完的镀银玻璃棒用去离子水冲洗并且在冷风中吹干备用。
[0100]
2、寡聚脱氧核糖核苷酸修饰的银板的制备:
[0101]
(1)将5nm的sw与7nm的bl混合,室温条件下反应30min,制备成swing arm-blocker,使sw完全与bl结合。bl过量能够保证sw的切割位点完成封闭,降低背景值;
[0102]
(2)将cp1及sd分别用10mmol/l pbs缓冲溶液(ph 7.0,0.1mol/l nano3)定容至体积为5.0ml,其中cp1与sd的摩尔比为1:10;
[0103]
(3)将上述制备得到的银板浸泡在1.1ml步骤(2)制备的溶液中,室温浸泡12h,将cp1及sd链修饰至银板上;然后,将银板抽出并用大量的pbs缓冲溶液冲洗银板以洗去银板表面未修饰的dna链;
[0104]
(4)再将上述步骤(3)中已修饰的银板浸泡在含有链霉亲和素(sa)溶液中,室温孵
育15分钟;将银板抽出并用大量的pbs缓冲溶液冲洗银板以洗去银板表面多余的sa;
[0105]
(5)再将上述步骤(4)修饰的银板浸泡在hrp-biotin溶液中,室温孵育15分钟,再将银板抽出并用大量的pbs缓冲溶液冲洗银板以洗去银板表面多余的hrp-biotin。
[0106]
实施例3双目标dna步行器的制备
[0107]
1、银板的制备:
[0108]
采用传统的银镜反应制得银纳米粒子覆盖的玻璃片(银板)。石英玻璃片以及所有使用的玻璃仪器均需氢氧化钠溶液,硝酸以及乙醇彻底清洗,然后用超纯水冲洗干净并吹干。
[0109]
首先制备银氨溶液,在洁净的烧杯中加入5ml的3%agno3溶液,然后加入1%nh3·
h2o,滴加的同时振荡,直至混合液澄清。然后将1cm
×
1cm的玻璃板放置于银氨溶液和10%的葡萄糖混合溶液当中(银氨溶液体积:葡萄糖溶液体积=1:2),反应在25℃下反应10分钟;最后将反应完的镀银玻璃棒用去离子水冲洗并且在冷风中吹干备用。
[0110]
2、寡聚脱氧核糖核苷酸修饰的银板的制备:
[0111]
(1)将5nm的sw与7nm的bl混合,室温条件下反应30min,制备成swing arm-blocker,使sw完全与bl结合。bl过量能够保证sw的切割位点完成封闭,降低背景值;
[0112]
(2)将cp1及sd分别用10mmol/l pbs缓冲溶液(ph 7.0,0.1mol/l nano3)定容至体积为5.0ml,其中cp1与sd的摩尔比为1:30;
[0113]
(3)将上述制备得到的银板浸泡在1.1ml步骤(2)制备的溶液中,室温浸泡12h,将cp1及sd链修饰至银板上;然后,将银板抽出并用大量的pbs缓冲溶液冲洗银板以洗去银板表面未修饰的dna链;
[0114]
(4)再将上述步骤(3)中已修饰的银板浸泡在含有链霉亲和素(sa)溶液中,室温孵育15分钟;将银板抽出并用大量的pbs缓冲溶液冲洗银板以洗去银板表面多余的sa;
[0115]
(5)再将上述步骤(4)修饰的银板浸泡在hrp-biotin溶液中,室温孵育15分钟,再将银板抽出并用大量的pbs缓冲溶液冲洗银板以洗去银板表面多余的hrp-biotin。
[0116]
实施例4双目标dna步行器的制备
[0117]
1、银板的制备:
[0118]
采用传统的银镜反应制得银纳米粒子覆盖的玻璃片(银板)。石英玻璃片以及所有使用的玻璃仪器均需氢氧化钠溶液,硝酸以及乙醇彻底清洗,然后用超纯水冲洗干净并吹干。
[0119]
首先制备银氨溶液,在洁净的烧杯中加入5ml的3%agno3溶液,然后加入1%nh3·
h2o,滴加的同时振荡,直至混合液澄清。然后将1cm
×
1cm的玻璃板放置于银氨溶液和10%的葡萄糖混合溶液当中(银氨溶液体积:葡萄糖溶液体积=1:2),反应在25℃下反应10分钟;最后将反应完的镀银玻璃棒用去离子水冲洗并且在冷风中吹干备用。
[0120]
2、寡聚脱氧核糖核苷酸修饰的银板的制备:
[0121]
(1)将5nm的sw与7nm的bl混合,室温条件下反应30min,制备成swing arm-blocker,使sw完全与bl结合。bl过量能够保证sw的切割位点完成封闭,降低背景值;
[0122]
(2)将cp1及sd分别用10mmol/l pbs缓冲溶液(ph 7.0,0.1mol/l nano3)定容至体积为5.0ml,其中cp1与sd的摩尔比为1:20;
[0123]
(3)将上述制备得到的银板浸泡在1.1ml步骤(2)制备的溶液中,室温浸泡12h,将
cp1及sd链修饰至银板上;然后,将银板抽出并用大量的pbs缓冲溶液冲洗银板以洗去银板表面未修饰的dna链;
[0124]
(4)再将上述步骤(3)中已修饰的银板浸泡在含有链霉亲和素(sa)溶液中,室温孵育15分钟;将银板抽出并用大量的pbs缓冲溶液冲洗银板以洗去银板表面多余的sa;
[0125]
(5)再将上述步骤(4)修饰的银板浸泡在hrp-biotin溶液中,室温孵育15分钟,再将银板抽出并用大量的pbs缓冲溶液冲洗银板以洗去银板表面多余的hrp-biotin。
[0126]
实施例5目标序列的检测
[0127]
采用实施例4中制备的已修饰的银板浸泡在含有t1、t2、4nm的sw、nb.bbvci酶及3nm cp 2溶液中,使总体系为1.1ml,室温杂交孵育45min。孵育后,抽出银板,并在余液中加入h2o2及120nmol/l abts
2-,室温孵育10min。最终溶液可通过紫外分光光度计在416nm处检测到信号值。
[0128]
双目标dna步行器的检测原理如图6所示:由于巯基(-sh)能够与金属银通过共价键链接,因此,cp1和sd能够联接到银板表面。cp1能够与t1中的14个碱基(第22844-22857位)杂交,从而实现特异性捕获t1。cp2能够与t1中的13个碱基杂交(第22828-22840位),还能与t2中的13个碱基(第22015-22027位)杂交,从而实现双目标dna的识别。sw能特异性识别t2的14个碱基(第21998-22011位),从而成功链接到银板上。由于修饰到银板上的sw与修饰在银板上的sd距离被大大缩短,通过精密的设计sw能够与sd形成类似分子内的关系,因此同一块银板上的sw与sd能够部分杂交形成双螺旋dna,并形成了nb.bbvci酶识别并切割的切割位点,将sd切割。
[0129]
同时,为了降低背景值,避免没联接到银板上的sw与sd结合。在反应前,用一段互补的封闭序列(bl)将sw上的切割位点封闭起来。当同一块银板上的sw与sd部分杂交形成双螺旋dna时,可将此段封闭序列置换出来。
[0130]
sd的5`端修饰巯基(-sh)用于联接银板,3`端修饰生物素(biotin)用于连接链霉亲和素(sa)。sa有四个结合位点,能够连接4个biotin分子。因此,将修饰了sd的银板泡入sa溶液能够将sa修饰在银板表面。再将银板取出,浸入辣根过氧化物酶-生物素(hrp-biotin)的溶液中,可以在sa的另外三个位点连上hrp-biotin。在hrp的催化作用下,abts
2-(无色)被h2o2氧化为abts
+
(绿色),溶液从无色变为绿色,作为信号输出。
[0131]
当体系中同时存在两段目标序列(t1和t2)时,由于双目标的识别,修饰在银板上的信号链(sd)与修饰在同一块银板上的游臂(sw)杂交形成双链dna,并产生了能被nb.bbvci酶识别并切割的切割位点,对sd的切割导致hrp从银板表面脱离掉落在溶液中。在加入h2o2及abts
2-后,abts
2-(无色)在hrp的催化下被h2o2氧化为abts
+
(绿色),溶液从无色变为绿色。最终反应溶液可通过紫外分光光度计在416nm处检测到信号值,并且,体系中的目标序列越多,信号值越大,溶液的颜色就越深,从而实现可视化检测目标序列的目的。
[0132]
实施例6双目标dna步行器的检测分析
[0133]
1、可行性分析
[0134]
为了检验本发明提供的双目标dna步行器检测的可行性,获取了采用双目标dna步行器对不同浓度的目标1、目标2存在情况的检测的信号反馈。因目标1、目标2为covid-19的s蛋白rna序列中的两部分不同序列片段,因此在选定检测浓度时,仅考虑同时不存在或等浓度存在,即可完成方法可行性验证,其他参数及条件全部同实施4和实施例5进行设置并
进行检测。
[0135]
不同浓度的目标1、目标2设置为:
[0136]
a组:0nmol/l的目标1和0nmol/l的目标2的混合目标;
[0137]
b组:300nmol/l的目标1和0nmol/l的目标2的混合目标;
[0138]
c组:0nmol/l的目标1和300nmol/l的目标2的混合目标;
[0139]
d组:300nmol/l的目标1和300nmol/l的目标2的混合目标。
[0140]
检测结果如图7所示,从吸光度上来看,d组的吸光度显著强于a,b,c组,即目标1和目标2同时存在时,体系的吸光度可达到0.695,而相反仅存在单独目标时或无目标时,吸光度趋近于0。同时从相应的实验反应图可以发现,只有在d组情况下,即目标1和目标2同时存在时,反应体系呈现出清晰的绿色。仅存在单独目标时或无目标时,并没有显著的变色现象。由此,该方法的可行性得到了证明。
[0141]
2、灵敏度分析
[0142]
为确定本发明提供的双目标dna步行器的灵敏度,将目标1和目标2分别取0-300nmol/l浓度并等量混合,其他参数及条件全部同实施4和实施例5进行设置并进行定量检测。
[0143]
结果如图8所示,双目标的浓度与abts
+
的紫外吸光度在5
×
10-9-3.0
×
10-7
mol/l范围内呈良好的线性关系,线性方程为:y=0.0277+0.00252x,检出限为1.19nmol/l,r2为0.995。
[0144]
3、特异性分析
[0145]
为验证本发明提供的双目标dna步行器的对t1、t2同检测择的特异性,使用本方法对含有单个及双个碱基错配的目标序列进行检测分析。检测目标设置为:单碱基错配的t1+t2目标混合物、双碱基错配的t1+t2目标混合物以及t1+t2目标混合物。碱基错配的t1、t2引物序列如下表2所示,上述目标物混合均以相对应的t1和t2以1:1等浓度混合进行前处理,浓度均选择300nmol/l,其他参数及条件全部同实施4和实施例5进行设置并进行检测。
[0146]
表2碱基错配的t1、t2引物序列
[0147][0148]
检测结果如图9所示,目标t1+t2的紫外吸收显著超过碱基错配的目标混合物的紫外吸收,对目标t1、t2表现出优异的选择性,证明了该方法对于t1、t2双目标识别的特异性。(n=3,mean
±
sd,**《0.001vs t1+t2)
[0149]
4、实验条件优化
[0150]
为优化本方法的检测性能,分别对捕获链1与信号链在银板上的修饰比例、体系反应时长以及abts
2-浓度进行优化。其他参数及条件全部同实施4和实施例5进行设置并进行检测。
[0151]
结果如图10~12所示,当sd与cp1在银板上的修饰比例为1:20,体系反应时长为45~75min,abts
2-浓度为100~150nmol/l时,该体系紫外吸收度最大,因此选择上述为该体系的最优条件。(n=3,mean
±
sd,**《0.001vs 1:20)
[0152]
实施例7实际样品的检测
[0153]
实验操作按照《新型冠状病毒实验室检测技术指南(第五版)》进行,咽拭子实际样品均先经过恒温培养箱60℃,30min灭活处理,与病毒保存液混合均匀待用。随后按照核酸提取试剂盒说明书,完成核酸提取,再通过rt-pcr试剂盒并按照说明书操作获取t1和t 2的单链rna片段。
[0154]
将获取的t1和t2以1:1等浓度混合,稀释至不同浓度用于检测(5nmol/l、25nmol/l、150nmol/l、300nmol/l),其他参数及条件全部同实施4和实施例5进行设置并进行检测。
[0155]
同时,在同等实验条件下,使用qubit
tm
rna(hs)检测试剂盒检测作为检测参照标准,对混合单链目标进行定量检测。并选用相对标准偏差对检测方法的精密度做以评估。所有测量均进行至少3次的平行检测实验以获取相应检测结果数据,并记录为平均值
±
标准差。所有数据均使用spss软件(23.0版;ibm)进行分析。多组数据间的统计学显著性用bonferroni的单因素方差分析进行事后检验。p《0.05时表明数据见差异具有统计学意义。
[0156]
检测结果如表3所示,本方法的平均回收率为95.8%-105.2%,相对标准偏差(rsd)为4.6%-6.5%。qubit
tm
rna(hs)定量试剂盒检测目标的平均回收率为92.5%-108.5%,相对标准偏差(rsd)为4.5%-9.2%。由此可证明本方法在实际样品检测中具有良好的精密度,且与试剂盒相比较具有更灵敏、更准确的优势。
[0157]
表3本发明与试剂盒检测方法对covid-19检测性能的比较
[0158][0159]
(n=3,平均值
±
标准差)
[0160]
本发明通过同时进行双目标检测,来启动dna步行器,当仅存在一个目标链或无目标时,不会触发dna步行器运行,无检测信号出现。当同时存在两个目标链时,捕获链cp1、cp2及sw特异性识别双靶标从而使步行器被目标链t1、t2激活,双靶标识别极大提升了covid-19识别的特异性和敏感度,降低检测过程中地假阳性干扰,防止对单一的目标检测出现的假阴性或者是假阳性结果,并提高双目标dna步行器的特异性和选择性,极大缩短检测时间、简化检出流程。
[0161]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,
均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1