杂环-三氮唑并噻二唑杂环串联化合物、合成方法、药物组合物及用途
1.本发明属于医药及其制备和应用的技术领域,具体涉及杂环-三氮唑并噻二唑杂环串联化合物、合成方法、药物组合物及用途。
背景技术:
2.shp2是一个在体内广泛存在的非受体型蛋白酪氨酸磷酸酶,由两个二个sh2结构域(n-sh2和c-sh2),一个具有催化活性的ptp结构域及富含脯氨酸基团及酪氨酸磷酸化尾巴组成。shp2作为血小板源性生长因子(pdgf)、表皮生长因子(egf)、成纤维细胞因子(fgf)、白细胞介素-3(il-3)、白血病抑制因子(lif)及α-干扰素(inf-α)等生长因子的下游信号分子,参与多条信号通路(例如ras/mark通路、pi3k/akt通路、jak/stat通路、jnk通路、nf-b通路、rho通路、nfat通路等),在细胞信息传递过程中起着关键的作用。shp2编码基因发生突变被认作是人类多种疾病的驱动力,例如在努南(noonan)综合征中有40-50%的患者发生了ptpn11的突变;在青少年粒单核细胞白血病(jmml)和急性髓细胞白血病(aml)中ptpn11的突变率分别达到35%和6.6%。在白血病中,shp2突变类型主要是e76k、d61y、e139d、q506p等,其中e76k这个突变类型是最常见的,也是与白血病最为密切的。因此,突变型shp2是潜在的抗肿瘤靶点。
3.近年来,shp2抑制剂取得了重要的进展。在发现第一个野生型shp2变构抑制剂shp099之后,出现了一些基于shp099结构改造的变构抑制剂,具体结构如下所示:
[0004][0005]
当前由各大制药公司开发出的shp2抑制剂如tno155、rmc-4630、jab-3068、et0038和rly-1971等抑制剂均处于临床研究当中。遗憾的是,现有的shp2抑制剂都不是突变型shp2抑制剂,不能满足临床药物开发的需求。因此,迫切需要发现更多结构新颖、选择性高的抑制剂,为研究突变型shp2在白血病信号通路中的生物功能提供工具化合物,为白血病治疗提供药物。
技术实现要素:
[0006]
本发明所要解决的技术问题是克服突变型shp2抑制剂的稀缺性问题,提供杂环-三氮唑并噻二唑杂环串联全新骨架类型的突变型shp2抑制剂、其中间体、合成方法、药物组合物及用途。该类化合物具有抑制蛋白酪氨酸磷酸酶shp2的生物活性,尤其对e76k突变型shp2具有高度选择性,在细胞中能有效抑制shp2下游信号通路的磷酸化水平,对肿瘤细胞具有很好的抑制活性,可以为预防和治疗癌症、代谢与免疫疾病提供新的手段,具有广阔的药物开发前景。
[0007]
本发明主要通过以下技术方案解决上述技术问题。
[0008]
[化合物]
[0009]
本发明提供了一种通式i所示的一类杂环-三氮唑并噻二唑杂环化合物或其药学上可接受的盐,
[0010][0011]
其中x独立选自o、-ch=ch-、-n=ch-,r1、r2分别独立选自-no2、-nh2、-sh、未取代和取代的芳香环、未取代和取代的杂芳香环、c
1-c6链烷基、环烷基、卤素、环氧烷基、烯基、炔基、醚链、-nhra、-shrb;其中ra选自氢、c
1-c6链烷基、环烷基、-c(o)-rc、未取代和取代的芳香环、未取代和取代的杂芳香环、未取代和取代的杂环烷基;rb选自氢、未取代和取代的芳香环、苯环上有取代或无取代的苄基;rc选自未取代和取代的芳香环、未取代和取代的杂芳香环。
[0012]
所述芳香环为苯环或者萘环。
[0013]
所述杂芳香环包括:呋喃环、噻吩环、哌啶环、吡啶环、吡咯环。
[0014]
所述杂环烷基为含有1-3个杂原子的c2-c5环烷基,杂原子包括s、n、o;具体可选:四氢呋喃环。
[0015]
所述取代的基团包括:卤素(f、cl、br、i)、c1-4烷基、c1-4烷氧基、-no2、-nhrd;rd选自氢、c
1-c6链烷基、环烷基、-c(o)-re、未取代和取代的芳香环、未取代和取代的杂芳香环、未取代和取代的杂环烷基;re选自未取代和取代的芳香环、未取代和取代的杂芳香环。
[0016]
优选地,当x为o时,为通式ⅱ,
[0017]
[0018]
r1,r2分别独立选自如下结构:nh2、sh、-cl、-f、-br、-i、-ch3、、
[0019]
优选地,x为-ch=ch-时,为式ⅲ所示结构,r1,r2分别独立选自如下结构:
[0020][0021]
r1,r2分别独立选自如下结构:-no2、-nh2、-sh、烯基、炔基、c
1-c6链烷基、醚链、含氧环烷基、环烷基、卤素、-nhra、-srb、未取代和取代的芳香环或杂芳香环、其中ra选自环丙基、环己基、异丙基、甲基,或者-nhra选自:
[0022]-srb选自:
[0023]
未取代和取代的芳香环为
[0024]
杂芳香环为
[0025]
优选地,当x为-n=ch-时,为式ⅳ所示结构,r1,r2分别独立选自如下结构:
[0026][0027]
r1,r2分别独立选自如下结构:-no2、-nh2、-sh、烯基、炔基、c
1-c6链烷基、醚链、含氧环烷基、环烷基、卤素、-nhra、-srb、未取代和取代的芳香环或杂芳香环、其中ra选自环丙基、环己基、异丙基、甲基,或者-nhra选自:
[0028]-srb选自:
[0029]
未取代和取代的芳香环为
[0030]
杂芳香环为
[0031]
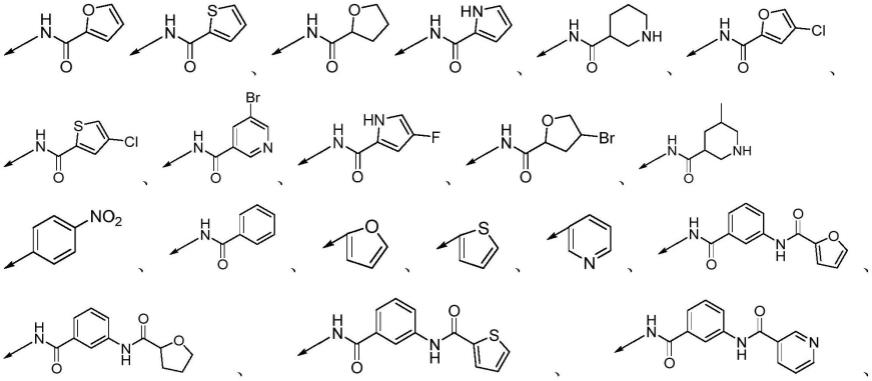
最优选地,一类杂环-三氮唑并噻二唑杂环化合物的具体结构为:
[0032]
[0033]
[0034]
[0035][0036]
[合成方法]
[0037]
本发明还提供了一种所述通式ⅰ化合物的合成方法,所述方法通过以下反应方案来实施:
[0038]
合成方案1
[0039][0040]
试剂和条件:a)二氯亚砜(socl2),无水甲醇,氮气(n2),80℃,12h;b)水合肼(n2h4.h2o),甲醇,80℃,12h;c)二硫化碳,氢氧化钾,无水甲醇,室温,12h;d)水合肼(n2h4.h2o),水,110℃,5h;e)溴化氰,75%乙醇,90℃,16h;f)2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu),n,n-二异丙基乙胺(dipea),n,n-二甲基甲酰胺(dmf),室温℃,2h;
[0041]
取i-1溶解于无水甲醇中,在冰浴条件下缓慢滴加二氯亚砜,n2保护后置于80℃油浴锅内回流14h,监测反应完全后,减压蒸馏除去meoh,固体加入乙酸乙酯和饱和碳酸氢钠溶液进行萃取,收集有机相,无水硫酸钠干燥,浓缩得化合物i-2,将i-2溶解于无水甲醇中,滴加水合肼,将该溶液置于80℃油浴锅内回流14h,监测反应完全后,减压蒸馏除去甲醇,固体用水和无水乙醇洗涤,真空干燥,得化合物i-3,将i-3溶解于无水甲醇中,将溶于无水甲醇的koh溶液缓慢滴加到上述溶液中搅拌,缓慢滴加cs2溶液,室温搅拌15h,监测反应完全后,抽滤固体,用无水乙醚进行洗涤,收集固体,直接投下一步,将i-4溶解在水中,滴加水合肼,将该溶液置于110℃油浴锅内加热回流3h,监测反应完全后,向反应液内加入冰水淬灭,在冰浴条件下用浓盐酸调ph 5-6,抽滤固体,真空干燥得i-5,将i-5,溴化氰溶解于75%乙醇溶液中,将反应液置于90℃油浴锅内加热回流20h,监测反应完全后,减压蒸馏至溶剂剩
1/4,加入饱和碳酸钠溶液,抽滤,得固体i-6,将i-6,i-7,hatu,dipea溶于dmf中,室温搅拌4h,监测反应完全后,将反应液滴入水中,析出固体,抽滤,经柱层析纯化得化合物i-8。
[0042]
合成方案2
[0043][0044]
试剂和条件:a)三氯氧磷,90℃,16h
[0045]
将ii-1,ii-2溶解在三氯氧磷中,90℃条件下加热回流16h,用水淬灭反应后加入50%氢氧化钠溶液调ph 7-8,用乙酸乙酯萃取后减压旋干溶剂,经柱层析纯化得化合物ii-3。
[0046]
合成方案3
[0047][0048]
试剂和反应条件:a)溴化亚铜(cubr2),亚硝酸叔丁酯(t-buno2),乙腈,0℃-室温,3h;b)碳酸铯,(4-甲基哌啶-4-基)氨基甲酸叔丁酯,n,n-二甲基甲酰胺(dmf),室温,12h;c)盐酸-1,4-二氧六环溶液(4m/l),室温,2-3h。
[0049]
将化合物iii-1和cubr2溶解于乙腈中,冰浴条件下缓慢滴加t-buno2,然后室温下反应3小时,反应完全后用etoac和1m/l稀盐酸洗涤,收集有机相,用无水na2so4干燥,减压旋蒸得化合物iii-2。将粗品iii-2、(4-甲基哌啶-4-基)氨基甲酸叔丁酯和碳酸铯加入溶剂dmf中,室温反应12小时后,将反应液滴入水中,用etoac萃取,有机相用饱和食盐水洗涤,将合并的有机相用无水na2so4干燥并减压旋蒸除去溶剂,粗品用柱层析纯化得化合物iii-3。将固体iii-3溶解在盐酸-1,4-二氧六环溶液(4m/l)中,室温反应2-3小时,抽滤固体,用甲基叔丁基醚(mtbe)和二氯甲烷(dcm)打浆,真空干燥得化合物iii-4。
[0050]
合成方案4
[0051][0052]
试剂和反应条件:a)氯乙酸,三氯氧磷,90℃,12h;b)碳酸铯,(4-甲基哌啶-4-基)氨基甲酸叔丁酯,dmf,室温,12h;c)盐酸-1,4-二氧六环溶液(4m/l),室温,2-3h。
[0053]
化合物iv-1和氯乙酸溶解在三氯氧磷中,90℃条件下加热回流12小时,将反应液缓慢滴加入水,用50%浓度的氢氧化钠中和至ph7-8,用etoac萃取,合并的有机相用无水na2so4干燥,旋干。粗品通过柱层析纯化得白色固体iv-2。将粗品iv-2、(4-甲基哌啶-4-基)氨基甲酸叔丁酯和碳酸铯加入溶剂dmf中,室温反应12小时后,将反应液滴入水中,用etoac萃取3次,用饱和食盐水洗涤1-2次,将合并的有机相用无水na2so4干燥并减压旋干溶剂,粗品用柱层析纯化得化合物iv-3。将固体iv-3溶解在盐酸-1,4-二氧六环溶液(4m/l)中,室温反应2-3小时,抽滤固体,用mtbe和dcm打浆,真空干燥得化合物iv-4。
[0054]
合成方案5
[0055][0056]
试剂和反应条件:a)二硫化碳,氢氧化钾,甲醇,90℃,24h;b)碳酸钾,乙腈,室温,12h。
[0057]
将v-1和氢氧化钾溶解在溶剂甲醇中,滴加二硫化碳溶液,将反应置于90℃油浴内加热回流24小时,监测反应完全后,用6m/l盐酸溶液调碱后经柱层析纯化得化合物v-2,将v-2,v-3和碳酸钾溶于乙腈中,室温反应12h,监测反应完全后抽滤,将滤液旋干后经柱层析纯化得化合物v-4。
[0058]
除特殊说明外,以上反应中所用试剂为本领域的常规试剂。例如,以上反应可以在如下溶剂中进行:n,n-二甲基甲酰胺(dmf)、乙醚、甲醇、水或上述溶剂的混合溶剂。根据具体化合物的反应情况,反应温度一般为室温或加热温度从45℃至110℃。反应时间根据具体反应物而定。所用缩合剂为本领域中常规的缩合剂,所用碱为本领域中常规的无机碱和有机碱,所用酯化试剂和还原试剂为本领域的常规酯化试剂和还原剂。通常用tlc来跟踪测定反应的完成程度,反应完毕后一般采用的后处理方法包括抽滤、浓缩反应液除尽溶剂、萃取、柱层析分离等。最终产物用nmr或者质谱来检测证明。
[0059]
[用途]
[0060]
通式i所示的化合物或其药学上可接受的盐在制备预防和治疗癌症、代谢与免疫疾病的药物中的用途。
[0061]
通式i所示的化合物或其药学上可接受的盐在制备蛋白酪氨酸磷酸酶shp2抑制剂中的用途。
[0062]
在所述用途中,通式i所示的化合物或其药学上可接受的盐在制备e76k突变在内的shp2获得性突变体、野生型shp2、shp1、tcptp以及ptp1b抑制剂中的应用。
[0063]
在所述用途中,通式i所示的化合物或其药学上可接受的盐在制备e76k突变在内的shp2获得性突变体、野生型shp2、shp1、tcptp以及ptp1b降解剂中的应用。
[0064]
[药物和药物组合物]
[0065]
本发明还提供了一种药物组合物,该组合物包含治疗有效量的所述通式i所示的化合物或其药学上可接受的盐,和任选的药学上可接受的辅料。其中,所述药物组合物用于预防和治疗癌症、代谢与免疫疾病。
[0066]
本发明还提供了一种用于预防和治疗癌症、代谢与免疫疾病、心血管病或者神经
性疾病的药物,所述药物包含如权利要求5中定义的通式i所示的化合物或其药学上可接受的盐,和药用辅料。
[0067]
所述辅料包含溶剂、抛射剂、增溶剂、助溶剂、乳化剂、着色剂、黏合剂、崩解剂、填充剂、润滑剂、润湿剂、渗透压调节剂、稳定剂、助流剂、矫味剂、防腐剂、助悬剂、包衣材料、芳香剂、抗黏合剂、整合剂、渗透促进剂、ph值调节剂、缓冲剂、增塑剂、表面活性剂、发泡剂、消泡剂、增稠剂、包合剂、保湿剂、吸收剂、稀释剂、絮凝剂与反絮凝剂、助滤剂以及释放阻滞剂。
[0068]
所述药物或者药物组合物还可以包括载体,所述载体包括微囊、微球、纳米粒和脂质体。
[0069]
所述药物的剂型包括注射液、注射用冻干粉针、控释注射剂、脂质体注射剂、混悬剂、植入剂、栓塞剂、胶囊剂、片剂、丸剂和口服液。
[0070]
有效效果:
[0071]
本发明含杂环-三氮唑并噻二唑杂环具有抑制蛋白酪氨酸磷酸酶shp2的生物活性,可以作为工具化合物研究蛋白酪氨酸磷酸酶shp2在细胞信号转导过程中的生物学功能关联性,为预防和治疗癌症、代谢与免疫疾病提供新的手段。
附图说明
[0072]
图1为化合物wj503抑制细胞增殖实验示意图。
[0073]
图2为化合物wj50的透析时间与shp2
e76k
活性关系示意。
[0074]
图3为化合物wj50对shp2
e76k
抑制效果示意图。
[0075]
图4为化合物wj503对shp2e76k抑制类型示意图。
具体实施方式
[0076]
本技术涉及的合成过程具备包括如下步骤:
[0077]
反应操作1:
[0078][0079]
试剂和条件:a)二氯亚砜(socl2),无水甲醇,氮气(n2),80℃,12h;b)水合肼(n2h4.h2o),甲醇,80℃,12h;c)二硫化碳,氢氧化钾,无水甲醇,室温,12h;d)水合肼(n2h4.h2o),水,110℃,5h;e)溴化氰,75%乙醇,90℃,16h;f)2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu),n,n-二异丙基乙胺(dipea),n,n-二甲基甲酰胺(dmf),室温℃,2h;
[0080]
取i-1溶解于无水甲醇中,在冰浴条件下缓慢滴加二氯亚砜,n2保护后置于80℃油浴锅内回流14h,监测反应完全后,减压蒸馏除去meoh,固体加入乙酸乙酯和饱和碳酸氢钠
溶液进行萃取,收集有机相,无水硫酸钠干燥,浓缩得化合物i-2,将i-2溶解于无水甲醇中,滴加水合肼,将该溶液置于80℃油浴锅内回流14h,监测反应完全后,减压蒸馏除去甲醇,固体用水和无水乙醇洗涤,真空干燥,得化合物i-3,将i-3溶解于无水甲醇中,将溶于无水甲醇的koh溶液缓慢滴加到上述溶液中搅拌,缓慢滴加cs2溶液,室温搅拌15h,监测反应完全后,抽滤固体,用无水乙醚进行洗涤,收集固体,直接投下一步,将i-4溶解在水中,滴加水合肼,将该溶液置于110℃油浴锅内加热回流3h,监测反应完全后,向反应液内加入冰水淬灭,在冰浴条件下用浓盐酸调ph 5-6,抽滤固体,真空干燥得i-5,将i-5,溴化氰溶解于75%乙醇溶液中,将反应液置于90℃油浴锅内加热回流20h,监测反应完全后,减压蒸馏至溶剂剩1/4,加入饱和碳酸钠溶液,抽滤,得固体i-6,将i-6,i-7,hatu,dipea溶于dmf中,室温搅拌4h,监测反应完全后,将反应液滴入水中,析出固体,抽滤,经柱层析纯化得化合物i-8。
[0081]
反应操作2
[0082][0083]
试剂和条件:a)三氯氧磷,90℃,16h
[0084]
将ii-1,ii-2溶解在三氯氧磷中,90℃条件下加热回流16h,用水淬灭反应后加入50%氢氧化钠溶液调ph 7-8,用乙酸乙酯萃取后减压旋干溶剂,经柱层析纯化得化合物ii-3。
[0085]
反应操作3
[0086][0087]
试剂和反应条件:a)溴化亚铜(cubr2),亚硝酸叔丁酯(t-buno2),乙腈,0℃-室温,3h;b)碳酸铯,(4-甲基哌啶-4-基)氨基甲酸叔丁酯,n,n-二甲基甲酰胺(dmf),室温,12h;c)盐酸-1,4-二氧六环溶液(4m/l),室温,2-3h
[0088]
将化合物iii-1和cubr2溶解于乙腈中,冰浴条件下缓慢滴加t-buno2,然后室温下反应3小时,反应完全后用etoac和1m/l稀盐酸洗涤,收集有机相,用无水na2so4干燥,减压旋蒸得化合物iii-2。将粗品iii-2、(4-甲基哌啶-4-基)氨基甲酸叔丁酯和碳酸铯加入溶剂dmf中,室温反应12小时后,将反应液滴入水中,用etoac萃取,有机相用饱和食盐水洗涤,将合并的有机相用无水na2so4干燥并减压旋蒸除去溶剂,粗品用柱层析纯化得化合物iii-3。将固体iii-3溶解在盐酸-1,4-二氧六环溶液(4m/l)中,室温反应2-3小时,抽滤固体,用甲基叔丁基醚(mtbe)和二氯甲烷(dcm)打浆,真空干燥得化合物iii-4。
[0089]
反应操作4
[0090][0091]
试剂和反应条件:a)氯乙酸,三氯氧磷,90℃,12h;b)碳酸铯,(4-甲基哌啶-4-基)氨基甲酸叔丁酯,dmf,室温,12h;c)盐酸-1,4-二氧六环溶液(4m/l),室温,2-3h
[0092]
化合物iv-1和氯乙酸溶解在三氯氧磷中,90℃条件下加热回流12小时,将反应液缓慢滴加入水,用50%浓度的氢氧化钠中和至ph7-8,用etoac萃取,合并的有机相用无水na2so4干燥,旋干。粗品通过柱层析纯化得白色固体iv-2。将粗品iv-2、(4-甲基哌啶-4-基)氨基甲酸叔丁酯和碳酸铯加入溶剂dmf中,室温反应12小时后,将反应液滴入水中,用etoac萃取3次,用饱和食盐水洗涤1-2次,将合并的有机相用无水na2so4干燥并减压旋干溶剂,粗品用柱层析纯化得化合物iv-3。将固体iv-3溶解在盐酸-1,4-二氧六环溶液(4m/l)中,室温反应2-3小时,抽滤固体,用mtbe和dcm打浆,真空干燥得化合物iv-4。
[0093]
反应操作5
[0094][0095]
试剂和反应条件:a)二硫化碳,氢氧化钾,甲醇,90℃,24h;b)碳酸钾,乙腈,室温,12h
[0096]
将v-1和氢氧化钾溶解在溶剂甲醇中,滴加二硫化碳溶液,将反应置于90℃油浴内加热回流24小时,监测反应完全后,用6m/l盐酸溶液调碱后经柱层析纯化得化合物v-2,将v-2,v-3和碳酸钾溶于乙腈中,室温反应12h,监测反应完全后抽滤,将滤液旋干后经柱层析纯化得化合物v-4。
[0097]
下述制备例中,1h-nmr谱采用bruker av
ⅲ‑
400mhz型核磁共振仪测定;质谱采用型质谱仪测定;试剂主要由上海化学试剂公司提供,产品纯化主要用柱层析法,硅胶(200-300目),柱色谱法所用的硅胶型号为粗空(zlx-ⅱ),由安徽良臣硅源材料有限公司生产。
[0098]
如未作特别说明,本发明所采用的方法和仪器等为本领域公知的技术。
[0099]
实施例1
[0100][0101]
试剂和条件:a)二氯亚砜(socl2),无水甲醇,氮气(n2),80℃,12h;b)水合肼(n2h4.h2o),甲醇,80℃,12h;c)二硫化碳,氢氧化钾,无水甲醇,室温,12h;d)水合肼(n2h4.h2o),水,110℃,5h;e)溴化氰,75%乙醇,90℃,16h;f)2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu),n,n-二异丙基乙胺(dipea),n,n-二甲基甲酰胺(dmf),室温℃,2h;
[0102]
取3-硝基-4甲氨基苯甲酸i-1(5.0g,0.025mol)溶解于50ml无水甲醇中,在冰浴条件下缓慢滴加二氯亚砜(6.1g,0.05mol),n2保护后置于80℃油浴锅内回流14h,监测反应完全后,减压蒸馏除去meoh,固体加入乙酸乙酯和饱和碳酸氢钠溶液进行萃取,收集有机相,无水硫酸钠干燥,浓缩得化合物i-2(3.7g,产率70%)
[0103]
称取i-2(3g,0.014mol)溶解于30ml无水甲醇中,滴加水合肼(3.6g,0.071mol),将该溶液置于80℃油浴锅内回流14h,监测反应完全后,减压蒸馏除去甲醇,固体用水和无水乙醇洗涤,真空干燥,得化合物i-3(2.1g,产率70%)
[0104]
称取i-3(2.0g,0.010mol)溶解于20ml无水甲醇中,将溶于无水甲醇的koh(0.841g,0.015mol)溶液缓慢滴加到上述溶液中搅拌,缓慢滴加cs2(1.142g,0.015mol)溶液,室温搅拌15h,监测反应完全后,抽滤固体,用无水乙醚进行洗涤,收集固体,直接投下一步。
[0105]
将i-4(2.0g,0.006mol)溶解在20ml水中,滴加水合肼(0.9g,0.018mol),将该溶液置于110℃油浴锅内加热回流3h,监测反应完全后,向反应液内加入30ml冰水淬灭,在冰浴条件下用浓盐酸调ph 5-6,抽滤固体,真空干燥得i-5(1.2g,产率70%)
[0106]
称取i-5(1g,0.004mol),溴化氰(0.530g,0.005mol)溶解于75%乙醇溶液中,将反应液置于90℃油浴锅内加热回流20h,监测反应完全后,减压蒸馏至溶剂剩1/4,加入饱和碳酸钠溶液,抽滤,得红色固体wj291(0.547g,产率50%)
[0107]
称取wj291(0.500g,0.002mol),2-呋喃甲酸i-7(0.224mol,0.002mol),hatu(0.760mg,0.002mol)溶于5ml dmf中,滴加dipea(0.774g,0.006mol),室温搅拌4h,监测反应完全后,将反应液滴入水中,析出固体,抽滤,得橙色固体wj385(0.385g,产率50%)。1h nmr(400mhz,dmso-d6)δ9.09(d,j=2.0hz,1h),8.48-8.45(q,j=4.8hz,1h),8.38(dd,j=9.2,2.0hz,1h),7.74(q,j=4.8hz,1h),7.21(d,j=9.2hz,1h),7.03(dd,j=3.2,0.8hz,
1h),6.56(q,j=1.6hz,1h),3.04(d,j=4.8hz,3h).ms(esi):m/z calcd for c
15h12
n7o4s[m+h]
+
386.1,found 386.0.
[0108]
实施例2
[0109][0110]
试剂和条件:a)三氯氧磷,90℃,16h;
[0111]
将ii-1(294.0mg,1.0mmol),ii-2(167.0mg,1.0mmol)溶解在三氯氧磷(5ml)中,90℃条件下加热回流16h,用水淬灭反应后加入50%氢氧化钠溶液调ph 7-8,用乙酸乙酯萃取后(30ml
×
2)减压旋干溶剂,经柱层析纯化得橙色固体wj425(207.0mg,产率:48.7%)。1h nmr(400mhz,dmso-d6)δ9.05(d,j=2.0hz,1h),8.49(d,j=8.4hz,2h),8.39(d,j=9.2hz,1h),8.30(d,j=8.4hz,2h),8.17(d,j=7.6hz,1h),7.38(d,j=9.2hz,1h),4.07
–
4.04(m,1h),1.34(d,j=6.0hz,6h).ms(esi):m/z calcd for c
18h16
n7o4s[m+h]
+
426.1,found 426.2.
[0112]
实施例3
[0113][0114]
试剂和反应条件:a)溴化亚铜(cubr2),亚硝酸叔丁酯(t-buno2),乙腈,0℃-室温,3h;b)碳酸铯,(4-甲基哌啶-4-基)氨基甲酸叔丁酯,dmf,室温,12h;c)盐酸-1,4-二氧六环溶液(4m/l),室温,2-3h;
[0115]
称取化合物wj284(511.2mg,1.8mmol)、cubr2(284.0mg,2.0mmol)溶于乙腈(8ml)中,冰浴条件下缓慢加入t-buno2(204.2mg,2.0mmol),缓慢升温至室温下反应3小时,反应完全后用etoac(20ml)和1m/l稀盐酸(10ml)洗涤,有机相用无水na2so4干燥,减压旋蒸除去溶剂得化合物iii-1(350.3mg,1.0mmol)。将粗品iii-1(300.0mg,0.9mmol)、(4-甲基哌啶-4-基)氨基甲酸叔丁酯(276.1mg,1.3mmol)和碳酸铯(559.0mg,1.7mmol)加入溶剂dmf(8ml)中,室温反应12小时后,将反应液滴入水中,用etoac萃取(20ml
×
2),用饱和食盐水(10ml
×
2)洗涤有机相,将合并的有机相用无水na2so4干燥并除去etoac,粗品用柱层析纯化得化合物wj482(285.1mg,0.6mmol)。将固体wj482溶解在5ml盐酸-1,4-二氧六环溶液(4m/l)中,室温反应2-3小时,抽滤固体,用mtbe和dcm淋洗,真空干燥得白色固体wj418(180.7mg,产率24.0%)。1h nmr(400mhz,dmso-d6)δ8.34(s,2h),7.87(t,j=8.0hz,1h),7.70(t,j=8.0hz,1h),7.57(t,j=8.0hz,1h),3.73
–
3.68(m,4h),1.90
–
1.78(m,4h),1.35-1.33(m,3h).ms(esi):m/z calcd for c
15h18
cl3n6s[m+h]
+
419.0,found 383.0.
[0116]
实施例4
[0117][0118]
试剂和反应条件:a)氯乙酸,三氯氧磷,90℃,12h;b)碳酸铯,(4-甲基哌啶-4-基)氨基甲酸叔丁酯,dmf,室温,12h;c)盐酸-1,4-二氧六环溶液(4m/l),室温,2-3h;
[0119]
化合物iv-1(984.2mg,3.8mmol)和氯乙酸(359.1mg,3.8mmol)溶解在三氯氧磷(12ml)中,90℃条件下加热回流12小时,将反应液缓慢滴加入水,用50%浓度的氢氧化钠中和至ph7-8,用etoac萃取(30ml
×
3),合并的有机相用无水na2so4干燥,旋干。粗品通过柱层析纯化得白色固体iv-2(540.6mg,1.7mmol)。将粗品iv-2(540.6mg,1.7mmol)、(4-甲基哌啶-4-基)氨基甲酸叔丁酯(545.7mg,2.6mmol)和碳酸铯(1.1g,3.4mmol)加入溶剂dmf(15ml)中,室温反应12小时后,将反应液滴入水中,用etoac萃取(20ml
×
3),用饱和食盐水洗涤有机相1次(20ml),将合并的有机相用无水na2so4干燥并减压旋蒸除去溶剂,粗品用柱层析纯化得化合物wj496(618.3mg,1.3mmol)。将固体wj496溶解在盐酸-1,4-二氧六环溶液(4m/l)(10ml)中,室温反应2-3小时,抽滤固体,用mtbe和dcm淋洗,真空干燥得化合物wj432(296.7mg,产率18.1%)。1h nmr(400mhz,dmso-d6)δ8.49(s,2h),7.92(d,j=7.6hz,1h),7.79(d,j=7.6hz,1h),7.61(t,j=7.6hz,1h),4.62(s,2h),3.41-3.40(m,2h),3.16-3.11(m,2h),2.08-2.02(m,2h),1.93-1.89(m,2h),1.36(s,3h),ms(esi):m/z calcd for c
16h20
cl3n6s[m+h]
+
433.1,found 397.0.
[0120]
实施例5
[0121][0122]
试剂和反应条件:a)二硫化碳,氢氧化钾,甲醇,90℃,24h;b)碳酸钾,乙腈,室温,12h;
[0123]
将v-1(259.0mg,1.0mmol)和氢氧化钾(56.1mg,1.0mmol)溶解在溶剂甲醇(15ml)中,滴加二硫化碳溶液(304.6mg,4.0mmol),将反应置于90℃油浴内加热回流24小时,监测反应完全后,用6m/l盐酸溶液调碱后经柱层析纯化得化合物wj301(100.0mg,0.3mmol),将wj301(80.0mg,0.27mmol),v-2(58.1mg,0.27mmol),碳酸钾(74.5mg,0.54mmol)溶解在溶剂乙腈中,室温反应12h,监测反应完全后,抽滤,滤液旋干就经柱层析纯化得白色固体wj436(50.0mg,产率:42.5%)。1h nmr(400mhz,dmso-d6)δ8.30(s,1h),8.15
–
8.13(m,1h),7.92(dd,j=8.0,1.6hz,1h),7.88(d,j=7.6hz,1h),7.73(dd,j=7.6,1.6hz,1h),7.61(q,j=8.0hz,2h),4.69(s,2h).ms(esi):m/z calcd for c
16h10
cl2n5o2s2[m+h]
+
438.0,found 438.0.
[0124]
除了适当替换相应的反应化合物外,以下化合物的制备参照上述制备的方法,得到不同化合物,结果如表1所示。
[0125]
表1不同杂环-三氮唑并噻二唑杂环化合物的表征数据结果
[0126]
[0127]
[0128]
[0129]
[0130]
[0131]
[0132]
[0133]
[0134][0135]
实验例6:含杂环-三氮唑并噻二唑杂环化合物抑制shp2活性测试
[0136]
1)材料:
[0137]
蛋白:shp2全长(met1-arg 593),将ptpn11基因克隆到含有n-末端6
×
his标签的pet-15b质粒中(cat.no.69661-3),通过大肠杆菌(bl21)表达系统表达得到his标签融合蛋白并借助akta avant25蛋白纯化系统进行分离和纯化。参考文献nature,2016,535(7610):148-152.
[0138]
2)过程:采用快速荧光定量检测法,在384孔黑色微孔微孔板(optiplate-384 black opaque,perkin elmer)中检测酶活性。底物difmup经shp2水解得到difmu并产生荧光。反应溶液体系为:60mm 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
model)(varible slope),对于大多数抑制剂筛选模型,将拟合曲线底部和顶部设定为0和100。一般情况下,每个样品在测试中均设置复孔(n≥3),在结果中以标准偏差(standard deviation,sd)或者标准误差(standard error,se)表示。每次测试均以shp099为参照(ic
50
=4.98
±
0.26μm)。所有数据都在我们知识能力范围内尽可能做到可信,精确,正确。
[0150]
实验例8:化合物抑制ptp结构域shp2活性测试
[0151]
应用大肠杆菌表达系统表达得到gst融合蛋白;荧光底物,omfp。过程:采用荧光底物omfp,在384黑底孔板中观察不同化合物对重组酶的活性的抑制。首先选取单点浓度50μm的化合物与酶在室温下孵育,最后迅速加入底物omfp,omfp水解底物omf在被485nm激发光激发后可发射出波长为530nm的可检测的荧光信号,从而观察酶的活性变化以及化合物对其的抑制情况。如果抑制率大于50%,则选取8个浓度,50μm为首要浓度的化合物做ic
50
测试。实验中采用的对照化合物为na3vo4。
[0152]
实验例9:化合物抑制野生型shp1活性测试
[0153]
应用大肠杆菌表达系统表达得到gst融合蛋白;荧光底物,omfp。过程:采用荧光底物omfp,观察不同化合物对重组酶的活性的抑制。首先选取单点浓度50μm的化合物与酶在室温下孵育,最后迅速加入底物omfp,omfp水解底物omf在被485nm激发光激发后可发射出波长为530nm的可检测的荧光信号,从而观察酶的活性变化以及化合物对其的抑制情况。如果抑制率(%inhibition)大于50%,则选取8个浓度,50μm为首要浓度的化合物做ic
50
测试
[0154]
实验例10:化合物抑制ptp结构域ptp1b活性测试
[0155]
应用大肠杆菌表达系统表达得到gst融合蛋白;荧光底物,omfp。过程:采用荧光底物omfp,在384黑底孔板中观察不同化合物对重组酶的活性的抑制。首先选取单点浓度50μm的化合物与酶在室温下孵育,最后迅速加入底物omfp,omfp水解底物omf在被485nm激发光激发后可发射出波长为530nm的可检测的荧光信号,从而观察酶的活性变化以及化合物对其的抑制情况。如果抑制率大于50%,则选取8个浓度,50μm为首要浓度的化合物做ic
50
测试。实验中采用的对照化合物为na3vo4。
[0156]
实验例11:化合物抑制ptp结构域tcptp活性测试
[0157]
应用大肠杆菌表达系统表达得到gst融合蛋白;底物,pnpp。过程:采用紫外底物pnpp,观察不同化合物对活性片断的活性抑制,以初步评价化合物的作用效果。tcptp水解底物pnpp的磷酯键得到的产物在405nm处有很强的光吸收。首先选取单点浓度50μm,2ml的化合物与88ml底物pnpp,直接加入10ml的ptp1b。因此可以直接监测405nm处光吸收的变化以观察酶的活性变化以及化合物对其的抑制情况。如果抑制率大于50%,则选取8个浓度,50μm为首要浓度的化合物做ic
50
测试。
[0158]
实验例12:化合物抑制mv-4-11细胞活性测试
[0159]
1)材料:
[0160]
细胞株:mv-4-11
[0161]
试剂:luminescent cell viability assay reagent细胞培养基:imdm,96孔白底板;
[0162]
2)过程:收集mv-4-11细胞,离心后重悬计数。将细胞以1.0
×
104个细胞/孔的密度接种于透明96孔细胞培养板中,每孔细胞悬液体积为80μl,将接种细胞后的培养板置于37℃恒温培养箱平衡至少30min,培养条件为37℃+5%co2。将化合物用培养基(imdm+10%
fbs)稀释至起始浓度500μm,2倍梯度稀释,并保证dmso浓度小于0.002%。将稀释好的化合物20μl加入细胞中,每个浓度3个复孔(化合物起始终浓度为100μm),在37℃恒温培养箱中培养7天。使用celltiteraqueous non-radioactive cell proliferation assay(mts)试剂检测细胞增殖情况,20μl/孔,37℃恒温培养箱中孵育2~3小时,置于酶标仪读值,读取双波长690nm及490nm,计算细胞相对活力。
[0163]
3)样品处理:样品用dmso溶解,-20℃保存,dmso在最终体系中的浓度控制在不影响检测活性的范围之内。
[0164]
4)数据处理及结果说明:
[0165]
测试活性剂量依赖关系,即ic
50
/ec
50
值,通过样品活性对样品浓度进行非线性拟和得到,计算所用软件为graphpad prism 6,拟合所使用的模型为四参数剂量效应积分模型(four-parameter concentration
–
response model)(varible slope),对于大多数抑制剂筛选模型,将拟合曲线底部和顶部设定为0和100。一般情况下,每个样品在测试中均设置复孔(n≥3),在结果中以标准偏差(standard deviation,sd)或者标准误差(standard error,se)表示。
[0166]
实施例13:化合物的酶动力学性质实验
[0167]
酶动力学实验技术:研究抑制剂的作用类型,可以进一步明确抑制剂的作用机理,推测抑制剂在酶上的结合部位及作用方式等。首先采用透析法或者大体积稀释法确定化合物是否是可逆抑制剂,如果是可逆抑制剂,则在不同的底物浓度(大约为酶的km值的1/8到8倍之间)下,检测反应初速度。然后根据michaelis-menten方程(公式1)拟合v~[s]曲线,得到km和vmax值。
[0168][0169]
可逆抑制剂又可以细分为竞争性抑制剂、非竞争性抑制剂及反竞争性抑制剂,根据km与vmax的关系,来具体判定是哪种类型的可逆抑制剂。除了做可逆抑制剂分析试验,同时还需要确定化合物与蛋白的结合快慢。
[0170]
所有数据都在我们知识能力范围内尽可能做到可信,精确,正确。
[0171]
所得测试结果见表2。
[0172]
表2含杂环-三氮唑并噻二唑杂环化合物的生物活性数据
[0173]
[0174]
[0175]
[0176]
[0177][0178]
其中,a代表ic
50
小于等于5μm,b代表5μm《ic
50
《20μm,c代表20μm《ic
50
《50μm,d代表ic
50
》50μm,
“‑”
代表活性未测。
[0179]
表2中阳性化合物wj503对mv-4-11细胞的抗增殖活性结果如图1所示。从图1可以看出化合物wj503(ic
50
=13.11
±
4.45μm)具有一定的抑制细胞生长活性,成剂量依赖型。
[0180]
表2中阳性化合物wj503的透析时间与shp2
e76k
活性关系如图2所示。图2结果显示,随着时间的延长,wj503逐步与shp2 e76k
解离,shp2 e76k
的活性逐步恢复,可以看出化合物wj503为可逆性抑制剂。
[0181]
表2中阳性化合物wj503对shp2
e76k
抑制效果与时间没有依赖关系如图3所示。图3结果显示,化合物wj503为快结合抑制剂。
[0182]
表2中阳性化合物wj503对shp2
e76k
抑制类型结果如图4所示。图4结果显示,化合物wj503表现出竞争型抑制剂特征。综上所述,化合物503为shp2
e76k
蛋白酶的一种可逆竞争型快结合抑制剂。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1