一种原位成核聚丙烯及其制备方法与流程
1.本发明属于石油化工技术领域,尤其涉及一种原位成核聚丙烯及其制备方法。
背景技术:
2.聚丙烯具有优异的物理力学性能及化学性能。然而,由于聚丙烯属于半结晶性聚合物,具有结晶速率慢、结晶度较低、在成型过程中易形成较大尺寸球晶等缺点,使其性能及应用受到了极大的限制,在聚丙烯中加入成核剂改善上述缺点的方法在本领域中是公知的。
3.原位成核技术中使用的成核剂又可以分为常规成核剂和聚合物成核剂。在常规成核剂原位成核技术研究中,捷克石油化工布尔诺(brno)聚合物研究所的专利us 2013/0190435a1、wo 2015/021948a1公开报道了通过烷基铝与脂肪族或芳香族羧酸酰胺衍生物的化合物反应制备的成核剂用于原位成核;中科院化学所的董金勇研究团队专利cn 102603941b报道了以常规β成核剂为载体的催化剂催化丙烯聚合,催化剂载体的作用确保了成核剂在聚丙烯基体中的良好分散;大连理工的牛慧研究团队将成熟的购买来的成核剂做成对催化剂惰性的成分,聚合时与催化剂一起加入,相较于传统加工成核法,成核剂添加量降低;华东理工大学的辛忠团队专利cn 1974611a报道了一种具有催化功能的聚烯烃用成核剂的制备方法及其应用;中国石油天然气股份有限公司专利cn 101735354b、cn 103524644b报道了在催化体系中加入稀土β型成核剂用于聚丙烯的原位成核。虽然,常规成核剂原位成核技术与传统加工成核技术相比,成核剂在500ppm以下就可以起到很好的成核效果,在聚合物中分散更加均匀,但是该技术需要对成核剂进行惰性处理,处理成本较高,同时降低了催化体系的催化活性。
4.在聚合物成核剂原位成核技术研究中,北欧化工原位成核技术中,在催化剂改性结束之后采用丙烯本体预聚合方式,原位聚合物成核剂浓度仍然在45~1000ppm;并且催化剂改性过程复杂,催化剂改性和主聚合反应不连续,会造成一定程度的催化剂损耗,从而影响体系催化活性。
技术实现要素:
5.有鉴于此,本发明的目的在于提供一种原位成核聚丙烯及其制备方法,该方法制备的原位成核聚丙烯具有较高结晶温度和结晶度。
6.本发明提供了一种原位成核聚丙烯的制备方法,包括以下步骤:
7.1)、将含钛主催化剂、助催化剂、成核剂单体加入反应釜内,再加入溶剂,反应,得到负载聚合物成核剂的催化剂;
8.2)、将助催化剂、外给电子体和负载聚合物成核剂的催化剂混合,通入气相丙烯,进行丙烯淤浆预聚合;
9.3)、预聚合结束后分离溶剂,再通液相丙烯和氢气进行本体聚合,得到原位成核聚丙烯。
10.在本发明中,所述主催化剂为氯化镁负载四氯化钛球形催化剂,即ziegler-natta催化剂;所述氯化镁负载四氯化钛球形催化剂选自n系列、dq系列、cs系列、stp系列和tk系列丙烯聚合催化剂中的一种或多种。
11.所述助催化剂为烷基铝化合物;所述烷基铝化合物选自三乙基铝、三异丁基铝、三己基铝、三辛基铝、二乙基一氯化铝、二异丁基一氯化铝、二己基一氯化铝和二辛基一氯化铝中的一种或多种;
12.所述成核剂单体选自乙烯基化合物;所述乙烯基化合物选自乙烯基环己烷、乙烯基环戊烷、乙烯基-2-甲基环己烷、3-甲基-1-丁烯、3-乙基-1-己烯、3-甲基-1-戊烯和苯乙烯中的一种或多种;
13.所述外给电子体选为硅烷化合物;所述硅烷化合物选自乙烯基三乙氧基硅烷、乙烯基三甲氧基硅烷、甲基三乙氧基硅烷、二甲基二甲氧基硅烷、二乙基二甲氧基硅烷、二丙基二甲氧基硅烷、二异丙基二甲氧基硅烷、二丁基二甲氧基硅烷、二异丁基二甲氧基硅烷、二叔丁基二甲氧基硅烷、二叔己基二甲氧基硅烷、二苯基二甲氧基硅烷、二环己基二甲氧基硅烷、二环戊基二甲氧基硅烷、二甲基二乙氧基硅烷、二乙基二乙氧基硅烷、二丙基二乙氧基硅烷、二异丙基二乙氧基硅烷、二丁基二乙氧基硅烷、二异丁基二乙氧基硅烷、二叔丁基二乙氧基硅烷、二叔己基二乙氧基硅烷、二苯基二乙氧基硅烷、二环己基二乙氧基硅烷、二环戊基二乙氧基硅烷、苯基三甲氧基硅烷、苯基三乙氧基硅烷、环戊基甲基二甲氧基硅烷、环戊基三甲氧基硅烷、环己基甲基二甲氧基硅烷、环己基三甲氧基硅烷、叔己基三甲氧基硅烷、叔丁基三甲氧基硅烷、叔己基三甲氧基硅烷中的一种或多种;
14.所述溶剂为烷烃;所述烷烃选自正戊烷、异戊烷、正已烷、正庚烷和正辛烷中的一种或多种。
15.在本发明中,在步骤1)反应过程中含钛主催化剂表面的四价钛被助催化剂还原为三价钛,参与成核剂单体的聚合反应;含钛主催化剂中被活化的钛含量占含钛主催化剂中总钛量的1~10%,优选为1~5%;反应得到的负载聚合物成核剂的催化剂的表面为聚合物成核剂;负载聚合物成核剂的催化剂中聚合物成核剂的质量占20~80%。
16.在本发明中,所述步骤1)中助催化剂和含钛主催化剂中钛的摩尔比为1~100:1;
17.所述步骤2)中助催化剂和步骤1)中含钛主催化剂中钛的摩尔比为20~300:1;
18.所述步骤2)中外给电子体和步骤1)中含钛主催化剂中钛的摩尔比为5~30:1;
19.所述成核剂单体和步骤1)中含钛主催化剂的质量比为0.5~5:1;
20.所述步骤1)中含钛主催化剂的质量与溶剂的体积比为(0.1~10)mg:1ml;
21.所述步骤2)中气相丙烯的压力为0.1~0.5mpa。
22.在本发明中,所述液相丙烯和步骤1)中含钛主催化剂中钛的摩尔比为1
×
105~1
×
107:1,氢气分压为0.5~5bar。
23.在本发明中,所述步骤1)反应的温度为0~50℃,反应的时间为5~20h;
24.所述步骤2)预聚合的温度为10~80℃,预聚合的时间为3~60min;
25.所述步骤3)本体聚合的温度为60~100℃,本体聚合的时间为1~5h。
26.本发明提供了一种原位成核聚丙烯,由上述技术方案所述制备方法制得;
27.所述原位成核聚丙烯的结晶温度高于128℃,结晶度大于55%;
28.所述原位成核聚丙烯中聚合物成核剂的浓度为20~30ppm。
29.本发明提供了一种原位成核聚丙烯的制备方法,包括以下步骤:将含钛主催化剂、助催化剂、成核剂单体加入反应釜内,再加入溶剂,反应,得到负载聚合物成核剂的催化剂;将助催化剂、外给电子体和负载聚合物成核剂的催化剂混合,通入气相丙烯,进行丙烯淤浆预聚合;预聚合结束后分离溶剂,再通液相丙烯和氢气进行本体聚合,得到原位成核聚丙烯。该方法一步法制备负载聚合物成核剂的催化剂后,直接通入气相丙烯进行淤浆预聚合,使催化剂颗粒充分破碎最大程度的释放活性中心以提高催化剂在丙烯本体聚合中的催化活性,最终制备的含低浓度聚合物成核剂的原位成核聚丙烯呈现出较高的结晶温度和结晶度。实验结果表明:所述原位成核聚丙烯的结晶温度高于128℃,结晶度大于55%;所述原位成核聚丙烯中聚合物成核剂的浓度为20~30ppm。
具体实施方式
30.为了进一步说明本发明,下面结合实施例对本发明提供的一种原位成核聚丙烯及其制备方法进行详细地描述,但不能将它们理解为对本发明保护范围的限定。
31.除非特别说明,本发明采用的方法和设备为本领域的常规方法和设备。
32.聚合物中聚乙烯基环己烷(pvch)的含量通过测试乙烯基环己烷(vch)的转化率计算。用气相色谱仪分析正已烷混合物中残留vch的量。甲苯用作内标。以下实施例和对比例中vch残余量均为0。
33.有关聚合物的测试条件如下:
34.熔体质量流动速率(mfr):按gb/t3682.1-2018测定,负荷2.16kg,测试温度230℃。
35.等规度(c7-ins):采用正庚烷抽提法测定(正庚烷沸腾抽提8h):将2g干燥的聚合物样品,放在索氏抽提器中用沸腾正庚烷抽提8h后,将剩余物干燥至恒重所得到的聚合物质量(g)与2(g)的比值即为等规度。
36.dsc测试:在氮气气氛,首先以10℃/min的速率将5~10mg样品从室温加热到200℃,并在200℃下保持5min以消除热历史;然后,以-10℃/min的速率将样品冷却至室温;最后,以10℃/min的速率将样品重新加热至200℃。根据以下关系式,使用第二次升温曲线计算样品的熔点(tc)和结晶度(xc),熔点(tm)也根据二次升温曲线测得:xc(%)=δhm/δh
*f
·
100%,其中,δhm是二次升温曲线的熔融焓,δh
*f
是聚丙烯的标准熔融焓(165.5j/g)
·
。
37.催化活性按以下公式计算:ca=q/w
ti
·
t*10-3
,kgpp
·
(gti
·
h)-1
,其中ca为催化剂催化活性,q为在聚合反应产物质量(g),w
ti
为催化剂钛用量(g),t为丙烯聚合时间(h)。
38.为了便于叙述,下列实施例中,所用的主催化剂为市售氯化镁负载四氯化钛型,如辽宁向阳科化集团催化剂公司的cs系列,钛含量为2.3wt%;助催化剂为三乙基铝,配成正已烷溶液,浓度为1mol/l;外给电子体为环己基甲基二甲氧基硅烷(donor-c)、二环戊基二甲氧基硅烷(donor-d)配成正庚烷溶液,浓度均为0.2mol/l;聚合物成核剂单体为乙烯基环己烷,密度0.805g/ml,纯度≥98%,水含量≤100ppm;正己烷已经过精制处理去除水分和杂质;丙烯聚合反应装置为3l高压聚合釜,其搅拌桨与聚合釜底间距1~3mm。
39.对比例1
40.3升的高压釜用高纯氮气充分置换后,依次将10mg主催化剂、1ml助催化剂、0.5ml外给电子体donor-c加入反应釜中,再加入100ml正已烷,并向反应釜中持续通入气相丙烯,
保持聚合釜压力在0.4mpa,在30℃下进行淤浆聚合10min;然后,在30℃下通过隔膜真空泵对反应釜抽真空将溶剂抽出分离,最后向反应釜内通入700g液相丙烯和1.5bar氢气进行本体聚合,在80℃下聚合2h。丙烯聚合及聚合物表征结果见表1。
41.对比例2
42.3升的高压釜用高纯氮气充分置换后,依次将10mg主催化剂、1ml助催化剂、0.5ml外给电子体donor-c加入反应釜中,再加700g液相丙烯,在30℃下进行本体预聚合10min;最后向反应釜内加入1.5bar氢气进行本体聚合,在80℃下聚合2h。丙烯聚合及聚合物表征结果见表1。
43.对比例3
44.正已烷中的催化剂的vch改性,如专利cn 109196003b。
45.将60ml正已烷、2ml助催化剂加入250ml玻璃反应釜中。20min后加入0.4g主催化剂,在室温下1h内加入0.6gvch,在50℃温度下聚合反应15h,冷却至室温。通过倾析出去混合物中的大部分正已烷,通过用氮气冲洗出去混合物中剩余的正已烷,得到改性后的固体催化剂0.95g,10mg主催化剂改性后成为23.75mg。
46.丙烯聚合:3升的高压釜用高纯氮气充分置换后,依次加入23.75mg改性后的主催化剂、0.95ml助催化剂、0.48ml外给电子体donor-c加入反应釜中,再加入100ml正已烷,并向反应釜中持续通入气相丙烯,保持聚合釜压力在0.4mpa,在30℃下进行淤浆聚合10min;然后,在30℃下通过隔膜真空泵对反应釜抽真空将溶剂抽出分离,最后向反应釜内通入700g液相丙烯和1.5bar氢气进行本体聚合,在80℃下聚合2h。丙烯聚合及聚合物表征结果见表1。
47.实施例1
48.3升的高压釜用高纯氮气充分置换后,依次加入10mg主催化剂、1ml助催化剂(原浓度稀释20倍至0.05mol/l)、15mgvch,再加入100ml正已烷,在30℃温度下聚合15h;聚合结束后将1ml助催化剂和0.5ml外给电子体donor-c加入反应釜中,再向反应釜中持续通入气相丙烯,保持聚合釜压力在0.4mpa,在30℃下进行淤浆预聚合10min;最后,在30℃下通过隔膜真空泵对反应釜抽真空将溶剂抽出分离,然后向反应釜内通入700g液相丙烯和1.5bar氢气进行本体聚合,在80℃下聚合2h。丙烯聚合及聚合物表征结果见表1。
49.实施例2
50.外给电子体选用donor-d,其他制备条件同实施例1。
51.实施例3
52.丙烯淤浆聚合为3min,其他制备条件同实施例1。
53.实施例4
54.丙烯淤浆聚合为6min,其他制备条件同实施例1。
55.实施利5
56.丙烯淤浆预聚合的时间为30min,其它制备条件同实施利1。
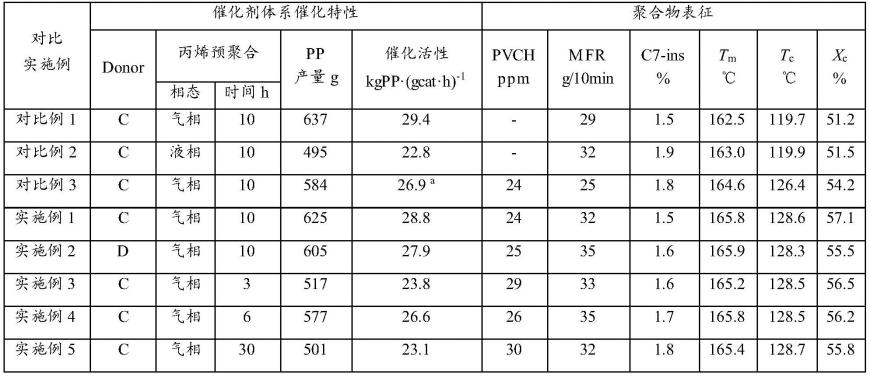
57.表1实施利和对比例的聚合物表征结果
[0058][0059]
注:a.对比例3按主催化剂用量10mg计算催化活性。
[0060]
催化活性是催化剂的重要性质之一,催化活性表示单位时间内单位催化剂活性组分的量所能产生的聚合物的质量,直接影响着产品的生产成本。从表1可以看出,相比于液相丙烯本体预聚合(对比例2),气相丙烯淤浆聚合体系(如实施例1~5)的催化活性更高,可以说明在相同的工艺条件下,丙烯气相聚合更有利于催化剂的破碎和活性中心的释放;实施例1与对比例1相比呈现出相近的催化活性,说明在相同的聚合工艺条件下,本发明的聚合物成核剂制备及添加对体系的催化活性基本没有影响;实施例1与对比例3相比催化活性更高,说明本发明的原位成核聚丙烯制备方法比前期专利报道的方法更有利于催化活性的保持和提高;实施例1、实施例3、实施例4和实施例5催化活性的不同可以说明,预聚合时间可以影响催化剂破碎及活性中心的释放,预聚合时间过段(3min、6min)催化剂破碎不完全,部分活性中心未释放,预聚合时间过长(30min),催化剂破碎释放的部分活性中心被聚丙烯所包埋,无法与丙烯单体接触进行聚合反应;实施例1和实施利2的对比说明,在使用不同的外给电子体条件下,均表现出很好的催化活性。
[0061]
结晶温度是成核剂效率的良好指标,较高的结晶温度意味着最终产品中更有效的成核。从表1中可以看出实施例1~5均呈现出了较好的成核效果,使聚丙烯的结晶温度从119.7~119.9℃提高到了128.3~128.7℃,且结晶度均大于55%;实施例1与对比例3(vch改性催化剂)在相同的丙烯聚合条件下,制备的原位成核聚丙烯中成核剂含量相近,但是本发明方法的成核效果更佳(128.6℃>126.4℃);从实施例1~5中可以看出,本发明方法制备的原位成核聚丙烯中成核剂浓度20~30ppm,成核效果却非常明显。
[0062]
由以上实施例可知,本发明提供了一种原位成核聚丙烯的制备方法,包括以下步骤:将含钛主催化剂、助催化剂、成核剂单体加入反应釜内,再加入溶剂,反应,得到负载聚合物成核剂的催化剂;将助催化剂、外给电子体和负载聚合物成核剂的催化剂混合,通入气相丙烯,进行丙烯淤浆预聚合;预聚合结束后分离溶剂,再通液相丙烯和氢气进行本体聚合,得到原位成核聚丙烯。该方法一步法制备聚合物成核剂后,直接通入气相丙烯进行淤浆预聚合,使催化剂颗粒充分破碎最大程度的释放活性中心以提高催化剂在丙烯本体聚合中的催化活性,最终制备的含低浓度聚合物成核剂的原位成核聚丙烯呈现出较高的结晶温度和结晶度。实验结果表明:所述原位成核聚丙烯的结晶温度高于128℃,结晶度大于55%;所述原位成核聚丙烯中聚合物成核剂的浓度为20~30ppm。
[0063]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人
员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1