一种三唑并吡啶酰胺类化合物及其制备方法和应用
1.本发明涉及农药技术领域,具体涉及一种三唑并吡啶酰胺类化合物及其制备方法和应用。
背景技术:
2.甲氧基丙烯酸酯类杀菌剂的活性基团是甲氧基丙烯酸(酯/酰胺),主要作用于真菌的线粒体呼吸链中的细胞色素bcl复合物,阻止电子传递从而抑制真菌生长,具有保护、治疗、铲除、渗透作用,无致癌和致突变等特点,能有效防治子囊菌、担子菌、半知菌和卵菌等真菌引起的病害,且具有高度的环境安全性,是目前应用最为广泛的植物病害用杀虫剂。然而,甲氧基丙烯酸酯类杀菌剂的抗药性已开始制约这类杀菌剂的进一步发展。
3.酰胺类杀菌剂可有效地克服甲氧基丙烯酸酯类杀菌剂的抗药性问题,是近年来研究十分活跃的杀菌剂品种之一。酰胺类杀菌剂的主要代表是琥珀酸脱氢酶抑制剂sdhis。伴随着sdhis类药剂的大量使用,田间已经有多种病菌对该类药剂产生了抗性。因此,发掘具有新结构的化学小分子农药为上述问题的解决提供了新途径。
技术实现要素:
4.有鉴于此,本发明的目的在于提供一种三唑并吡啶酰胺类化合物及其制备方法和应用,本发明提供的三唑并吡啶酰胺类化合物对植物真菌病害具有强烈的抑制作用。
5.为了实现上述发明目的,本发明提供以下技术方案:
6.本发明提供了一种三唑并吡啶酰胺类化合物,具有式i所示的结构:
[0007][0008]
式i中,所述x包括氧或硫;
[0009]
所述r1和r2独立地包括氢、c1~c6烷基、未取代或取代的苯基、未取代或取代的杂环基;r1和r2的碳手性中心的立体构型独立地为r型或s型;当r1和r2均不为氢时,*表示手性中心;
[0010]
所述r3包括氢、羟基、氨基、卤素、c1~c6烷基、c1~c6烷氧基、c1~c6烷胺基或c1~c6卤代烷基;所述r3的个数为1~3个;
[0011]
所述r4包括氢、c1~c6烷基、环己基、烷基羰基、c1~c6烷氧基、c1~c6卤代烷基、未取代或取代的苯基、未取代或取代的杂环基;所述r4的个数为1~3个;
[0012]
所述r5包括氢、c1~c6烷基、c1~c6烷氧基、未取代或取代的苯基、未取代或取代
的杂环基。
[0013]
优选的,r1、r2、r4和r5中,
[0014]
所述取代的苯基中的取代基包括苯基、卤素、羟基、氨基、c1~c6烷基、c1~c6烷氧基、c1~c6烷胺基或c1~c6卤代烷基;
[0015]
所述取代的杂环基中的取代基包括杂环基、卤素、羟基、氨基、c1~c6烷氧基、c1~c6烷胺基或c1~c6卤代烷基;所述未取代或取代的杂环基中的杂环基包括吡啶基、噻吩基或呋喃基。
[0016]
优选的,所述三唑并吡啶酰胺类化合物具有式i-1~i-8任一所示的结构:
[0017]
本发明提供了上述技术方案所述三唑并吡啶酰胺类化合物的制备方法,包括以下步骤:
[0018]
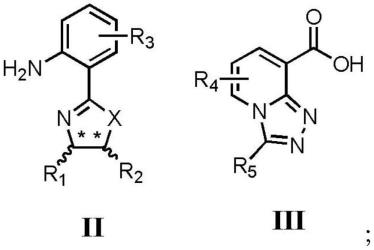
将化合物ii、化合物iii、缩合试剂、活化剂和有机溶剂混合,进行缩合反应,得到所述三唑并吡啶酰胺类化合物;
[0019][0020]
其中,r1~r5与所述式i中相同。
[0021]
优选的,所述有机溶剂包括卤代烷类溶剂、杂环类溶剂和苯类溶剂中的一种或几种;
[0022]
所述缩合反应的温度为0~40℃,时间为12~96h。
[0023]
优选的,所述化合物ii包括化合物ii-1或化合物ii-2;
[0024]
所述化合物ii-1的制备方法包括以下步骤:
[0025]
将化合物1、化合物2、催化剂和苯类溶剂混合,进行witte seeliger环化反应,得到化合物ii-1;
[0026]
所述化合物ii-2的制备方法包括以下步骤:
[0027]
将所述化合物ii-1、五硫化二磷和苯类溶剂混合,进行取代反应,得到化合物ii-2;
[0028][0029]
其中,r1~r3与所述式i中相同。
[0030]
优选的,所述化合物iii的制备方法包括以下步骤:
[0031]
将化合物3、水合肼和杂环类溶剂混合,进行取代反应,得到化合物4;
[0032]
当r5为氢时,将所述化合物4与原甲酸三酯混合进行环化反应,得到化合物6;
[0033]
当r5为其他取代基时,将所述化合物4、化合物5、碱金属盐、乙酸和有机溶剂混合,进行环化反应,得到化合物6;
[0034]
将所述化合物6、碱性试剂和醇类溶剂混合进行水解反应,得到化合物iii;
[0035][0036]
其中,r4和r5与所述式i中相同。
[0037]
本发明提供了上述技术方案所述的三唑并吡啶酰胺类化合物或上述技术方案所述制备方法得到的三唑并吡啶酰胺类化合物在制备抗植物病原真菌药物中的应用。
[0038]
优选的,所述植物病原真菌包括水稻纹枯病菌、小麦纹枯病菌、油菜菌核病菌、小麦赤霉病菌、小麦全蚀病菌、番茄灰霉病菌、马铃薯晚疫病菌、辣椒疫霉病菌、番茄早疫病菌、水稻恶苗病菌、马铃薯干腐病菌、黄瓜炭疽病菌和水稻稻瘟病菌中的一种或几种。
[0039]
本发明提供了一种抗植物病原真菌药物,包括活性组分和药用辅料;所述活性组分包括上述技术方案所述的三唑并吡啶酰胺类化合物或上述技术方案所述制备方法得到的三唑并吡啶酰胺类化合物。
[0040]
本发明提供了一种三唑并吡啶酰胺类化合物,具有式i所示的结构;式i中,所述x包括氧或硫;所述r1和r2独立地包括氢、c1~c6烷基、未取代或取代的苯基、未取代或取代的杂环基;r1和r2的碳手性中心的立体构型独立地为r型或s型;所述r3包括氢、羟基、氨基、卤素、c1~c6烷基、c1~c6烷氧基、c1~c6烷胺基或c1~c6卤代烷基;所述r3的个数为1~3个;所述r4包括氢、c1~c6烷基、环己基、烷基羰基、c1~c6烷氧基、c1~c6卤代烷基、未取代或取代的苯基、未取代或取代的杂环基;所述r5包括氢、c1~c6烷基、c1~c6烷氧基、未取代或取代的苯基、未取代或取代的杂环基。本发明提供三唑并吡啶酰胺类化合物,三唑并吡啶酰胺类新骨架具有抑菌活性,并在此基础上进行结构修饰和优化,获得了结构新颖的三唑并吡啶酰胺类杀菌剂候选化合物;本发明提供的三唑并吡啶酰胺类化合物对水稻纹枯病菌、小麦纹枯病菌、油菜菌核病菌、小麦赤霉病菌、小麦全蚀病菌、番茄灰霉病菌、马铃薯晚疫病菌、辣椒疫霉病菌、番茄早疫病菌、水稻恶苗病菌、马铃薯干腐病菌、黄瓜炭疽病菌和水稻稻瘟病菌具有很好的抑菌活性,在制备抗植物病原真菌药物中具有很好的应用前景。
[0041]
本发明提供了上述技术方案所述三唑并吡啶酰胺类化合物的制备方法。本发明提供的制备方法操作简单,制备原料成本低,绿色环保,适用工业化生产。
具体实施方式
[0042]
本发明提供了一种三唑并吡啶酰胺类化合物,具有式i所示的结构:
[0043][0044]
在本发明中,所述式i中x包括氧或硫。在本发明中,所述r1和r2独立地包括氢、c1~c6烷基、未取代或取代的苯基、未取代或取代的杂环基。在本发明中,所述c1~c6烷基优选包括氢、甲基、乙基、正丙基、异丙基、正丁基、仲丁基或叔丁基。在本发明中,所述取代的苯基中的取代基优选包括苯基、卤素、羟基(-oh)、氨基(-nh2)、c1~c6烷基、c1~c6烷氧基、c1~c6烷胺基、c1~c6卤代烷基。在本发明中,所述卤素优选为氟、氯、溴或碘。在本发明中,所述c1~c6烷氧基优选包括-och3、-och2ch3、-o(ch2)2ch3、-och(ch3)2、-o(ch2)3ch3、-och2ch(ch3)2、-ochch3ch2ch3、-o(ch3)3、-o(ch2)4ch3、-och(ch3)ch2ch2ch3、-och2ch(ch3)ch2ch3、-och2ch2ch(ch3)2、-och(ch3)2ch2ch3或-och2c(ch3)3。在本发明中,所述c1~c6烷胺基优选包括-nhch3、-nhch2ch3、-nh(ch2)2ch3、-nhch(ch3)2、-nh(ch2)3ch3、-nhch2ch(ch3)2、-nhch(ch3)ch2ch3、-nh(ch3)3、-nh(ch2)4ch3、-nhch(ch3)ch2ch2ch3、-nhch2ch(ch3)ch2ch3、-nhch2ch2ch(ch3)2、-nhc(ch3)2ch2ch3或-nhch2c(ch3)3。在本发明中,所述c1~c6卤代烷基优选为卤代甲基,所述卤代甲基中的卤素优选为氟、氯、溴或碘;所述卤代甲基优选为-ch2cl或-cf3。在本发明中,所述未取代或取代的杂环基中的杂环基优选包括吡啶基、噻吩基或呋喃基;所述取代的杂环基中的取代基优选包括杂环基、卤素、羟基、氨基、c1~c6烷氧基、c1~c6烷胺基或c1~c6卤代烷基;所述杂环基优选包括吡啶基、噻吩基或呋喃基;所述卤素、c1~c6烷氧基、c1~c6烷胺基或和c1~c6卤代烷基的可选取代基种类与前述卤素、c1~c6烷氧基、c1~c6烷胺基或和c1~c6卤代烷基的可选取代基种类相同,在此不再一一赘述。在本发明中,所述r1和r2的碳手性中心的立体构型独立地为r型或s型;当r1和r2均不为氢时,*表示手性中心。
[0045]
在本发明中,所述式i中r3包括氢、羟基、氨基、卤素、c1~c6烷基、c1~c6烷氧基、c1~c6烷胺基或c1~c6卤代烷基;所述r3的个数为1~3个,具体为1、2或3。在本发明中,所述卤素、c1~c6烷基、c1~c6烷氧基、c1~c6烷胺基和c1~c6卤代烷基的可选取代基种类与前述卤素、c1~c6烷氧基、c1~c6烷胺基或和c1~c6卤代烷基的可选取代基种类相同,在此不再一一赘述。
[0046]
在本发明中,所述式i中r4包括氢、c1~c6烷基、环己基、烷基羰基、c1~c6烷氧基、c1~c6卤代烷基、未取代或取代的苯基、未取代或取代的杂环基。在本发明中,所述烷基羰基优选包括甲基羰基、乙基羰基、正丙基羰基、异丙基羰基、正丁基羰基、仲丁基羰基、叔丁基羰基或苄基羰基。在本发明中,所述c1~c6烷基、c1~c6烷氧基、c1~c6卤代烷基、未取代或取代的苯基、未取代或取代的杂环基的可选取代基种类与前述c1~c6烷基、c1~c6烷氧
基、c1~c6卤代烷基、未取代或取代的苯基、未取代或取代的杂环基的可选取代基种类相同,在此不再一一赘述。
[0047]
在本发明中,所述式i中r5包括氢、c1~c6烷基、c1~c6烷氧基、未取代或取代的苯基、未取代或取代的杂环基。在本发明中,所述c1~c6烷基、c1~c6烷氧基、未取代或取代的苯基、未取代或取代的杂环基的可选取代基种类与前述c1~c6烷基、c1~c6烷氧基、未取代或取代的苯基、未取代或取代的杂环基的可选取代基种类相同,在此不再一一赘述。
[0048]
在本发明中,所述三唑并吡啶酰胺类化合物优选具有式i-1~i-8任一所示的结构:
[0049][0050]
本发明提供了上述技术方案所述三唑并吡啶酰胺类化合物的制备方法,包括以下步骤:
[0051]
将化合物ii、化合物iii、缩合试剂、活化剂和有机溶剂混合,进行缩合反应,得到所述三唑并吡啶酰胺类化合物;
[0052][0053]
其中,r1~r5与所述式i中相同。
[0054]
在本发明中,若无特殊说明,所有的原料组分均为本领域技术人员熟知的市售商品。
[0055]
在本发明中,所述化合物ii包括化合物ii-1或化合物ii-2。在本发明中,所述化合物ii的制备路线如下:
[0056][0057]
其中,r1~r3与所述式i中相同。
[0058]
在本发明中,所述所述化合物ii-1的制备方法优选包括以下步骤:将化合物1、化合物2、催化剂和苯类溶剂混合,进行witte seeliger环化反应,得到化合物ii-1。在本发明中,所述化合物1与化合物2的摩尔比优选1:1~1.5,更优选为1:1.1~1.4,进一步优选为1:1.2~1.3。在本发明中,所述催化剂优选包括锌盐,所述锌盐优选包括氯化锌、氟化锌、溴化锌和醋酸锌中的一种或几种;所述锌盐优选为无水锌盐。在本发明中,所述化合物1与催化剂的摩尔比优选1:0.5~3,更优选为1:1~2.5,进一步优选为1:1.5~2。在本发明中,所述苯类溶剂优选包括氯苯、甲苯、二甲苯、氟苯和溴苯中的一种或几种;所述苯类溶剂优选为无水苯类溶剂;本发明对于所述苯类溶剂的用量没有特殊限定,能够使得witte seeliger环化反应顺利进行即可。本发明对于所述混合没有特殊限定,能够将原料混合均匀即可,具体如搅拌混合。在本发明中,所述witte seeliger环化反应的温度优选为100~150℃,更优选为110~140℃,进一步优选为120~130℃;所述witte seeliger环化反应的时间优选为12~96h,更优选为20~80h,进一步优选为30~50h。完成witte seeliger环化反应后,本发明优选还包括后处理,所述后处理优选包括:将所得环化反应液浓缩,加入淬灭剂淬灭反应,有机溶剂萃取,将所得有机相进行干燥剂干燥,柱层析分离,得到化合物ii-1。本发明对于所述浓缩没有特殊限定,采用本领域技术人员熟知的浓缩方式即可,具体如减压蒸馏,本发明对于所述减压蒸馏没有特殊限定,能够将溶剂除去即可。在本发明中,所述淬灭剂优选包括氢氧化物水溶液,所述氢氧化物溶液的质量浓度优选0~35%,更优选为10~20%;所述化合物1与淬灭剂的用量比优选为1mmol:0.2~1ml,更优选为1mmol:0.5~0.8ml。在本发明中,所述萃取用有机溶剂优选包括乙酸乙酯或二氯甲烷。在本发明中,所述干燥剂优选包括无水硫酸钠。在本发明中,所述柱层析分离优选利用二氧化硅柱层析硅胶柱进行梯度洗脱,洗脱剂优选包括乙酸乙酯-石油醚混合溶剂,所述乙酸乙酯-石油醚混合溶剂中乙酸乙酯与石油醚的体积比优选为1:1~15。
[0059]
在本发明中,所述化合物ii-2的制备方法优选包括以下步骤:将所述化合物ii-1、五硫化二磷和苯类溶剂混合,进行取代反应,得到化合物ii-2。在本发明中,所述化合物ii-1与五硫化二磷的的摩尔比优选1:1.5~3,更优选为1:2~2.5。在本发明中,所述苯类溶剂优选包括甲苯和二甲苯中的一种或几种;所述苯类溶剂优选为无水苯类溶剂;本发明对于所述苯类溶剂的用量没有特殊限定,能够使得取代反应顺利进行即可。本发明对于所述混合没有特殊限定,能够将原料混合均匀即可,具体如搅拌混合。在本发明中,所述取代反应的温度优选为90~110℃,更优选为95~105℃,进一步优选为100℃;所述取代反应的时间优选为6~72h,更优选为20~60h,进一步优选为30~50h。完成取代反应后,本发明优选还包括后处理,所述后处理优选包括:将所得取代反应液冷却至室温,加入淬灭剂淬灭反应,
固液分离,将所得液体组分分相,将所得有机相进行干燥剂干燥,浓缩后柱层析分离,得到化合物ii-2。本发明对于所述冷却没有特殊限定,采用本领域技术人员熟知的冷却方式即可,具体如自然冷却。在本发明中,所述淬灭剂优选包括碱性水溶液,更优选包括naoh溶液、nahco3溶液和na2co3溶液中的一种或几种,所述碱性水溶液的质量浓度优选5~60%,更优选为20~40%;所述化合物1与淬灭剂的用量比优选为1mmol:0.4~1ml,更优选为1mmol:0.5~0.8ml。本发明对于所述固液分离没有特殊限定,采用本领域技术人员熟知的固液分离方式即可,具体如离心分离、过滤或抽滤。在本发明中,所述分相优选利用分液漏斗进行。在本发明中,所述干燥剂优选包括无水硫酸钠。本发明对于所述浓缩没有特殊限定,采用本领域技术人员熟知的浓缩方式即可,具体如减压蒸馏,本发明对于所述减压蒸馏没有特殊限定,能够将溶剂除去即可。在本发明中,所述柱层析分离优选利用二氧化硅柱层析硅胶柱进行梯度洗脱,洗脱剂优选包括乙酸乙酯-石油醚混合溶剂,所述乙酸乙酯-石油醚混合溶剂中乙酸乙酯与石油醚的体积比优选为1:1~15。
[0060]
在本发明中,所述化合物iii的制备方法优选包括以下步骤:
[0061]
将化合物3、水合肼和杂环类溶剂混合,进行取代反应,得到化合物4;
[0062]
当r5为氢时,将所述化合物4与原甲酸三酯混合进行环化反应,得到化合物6;
[0063]
当r5为其他取代基时,将所述化合物4、化合物5、碱金属盐、乙酸和有机溶剂混合,进行环化反应,得到化合物6;
[0064]
将所述化合物6、碱性试剂和醇类溶剂混合进行水解反应,得到化合物iii;
[0065][0066]
其中,r4和r5与所述式i中相同。
[0067]
在本发明中,所述化合物iii的制备路线如下:
[0068][0069]
本发明将化合物3、水合肼和杂环类溶剂混合,进行取代反应,得到化合物4。在本发明中,所述水合肼的质量浓度优选为40~81%,更优选为50~81%;所述化合物3与水合肼的摩尔比优选为1:1~2,更优选为1:1.2~1.8,进一步优选为1:1.4~1.5。在本发明中,所述杂环类溶剂优选包括二氧六环和/或四氢呋喃;本发明对于所述杂环类溶剂的用量没有特殊限定,能够使得取代反应顺利进行即可。本发明对于所述混合没有特殊限定,能够将
原料混合均匀即可,具体如搅拌混合。在本发明中,所述取代反应的温度优选为0~100℃,更优选为20~80℃,进一步优选为50~60℃;所述取代反应的时间优选为2~24h,更优选为3~15h,进一步优选为4~8h。完成取代反应后,本发明优选还包括后处理,所述后处理优选包括:将所得取代反应液冷却至室温,浓缩后柱层析分离,得到化合物4。本发明对于所述冷却没有特殊限定,采用本领域技术人员熟知的冷却方式即可,具体如自然冷却。本发明对于所述浓缩没有特殊限定,采用本领域技术人员熟知的浓缩方式即可,具体如减压蒸馏,本发明对于所述减压蒸馏没有特殊限定,能够将溶剂除去即可。在本发明中,所述柱层析分离优选利用二氧化硅柱层析硅胶柱进行梯度洗脱,洗脱剂优选包括乙酸乙酯-石油醚混合溶剂,所述乙酸乙酯-石油醚混合溶剂中乙酸乙酯与石油醚的体积比优选为1:0.5~5。
[0070]
得到化合物4后,当r5为氢时,本发明将所述化合物4与原甲酸三酯混合进行环化反应,得到化合物6。在本发明中,所述原甲酸三酯优选包括原甲酸三甲酯和/或原甲酸三乙酯。在本发明中,所述化合物4与原甲酸三酯的摩尔比优选为1:2~10,更优选为1:5~8。本发明对于所述混合没有特殊限定,能够将原料混合均匀即可,具体如搅拌混合。在本发明中,所述环化反应的温度优选为60~100℃,更优选为70~100℃,进一步优选为80~100℃;所述环化反应的时间优选为12~72h,更优选为20~60h,进一步优选为30~50h。完成环化反应后,本发明优选还包括后处理,所述后处理优选包括:将所得环化反应液冷却至室温,浓缩后柱层析分离,得到化合物6。本发明对于所述冷却没有特殊限定,采用本领域技术人员熟知的冷却方式即可,具体如自然冷却。本发明对于所述浓缩没有特殊限定,采用本领域技术人员熟知的浓缩方式即可,具体如减压蒸馏,本发明对于所述减压蒸馏没有特殊限定,能够将溶剂除去即可。在本发明中,所述柱层析分离优选利用二氧化硅柱层析硅胶柱进行梯度洗脱,洗脱剂优选包括乙酸乙酯-甲醇混合溶剂,所述乙酸乙酯-甲醇混合溶剂中甲醇与乙酸乙酯的体积比优选为1:10~20。
[0071]
得到化合物4后,当r5为其他取代基时,本发明将所述化合物4、化合物5、碱金属盐、乙酸和有机溶剂混合,进行环化反应,得到化合物6。在本发明中,所述碱金属盐优选包括乙酸钠和/或碳酸氢钠。在本发明中,所述化合物4、化合物5、碱金属盐和乙酸的摩尔比优选为1:0.5~5:0.5~5:0.5~5,更优选为1:0.5~3:0.5~3:0.5~3,进一步优选为1:1~2:1~2:1~2;在本发明中,所述有机溶剂优选包括醇类溶剂、杂环类溶剂和苯类溶剂中的一种或几种,更优选包括乙醇、甲醇、四氢呋喃、二氧六环、甲苯和氯苯的一种或几种,所述有机溶剂优选为无水有机溶剂;本发明对于所述有机溶剂的用量没有特殊限定,能够使得环化反应顺利进行即可。本发明对于所述混合没有特殊限定,能够将原料混合均匀即可,具体如搅拌混合。在本发明中,所述环化反应的温度优选为30~60℃,更优选为40~50℃;所述环化反应的时间优选为6~72h,更优选为10~60h,进一步优选为30~50h。完成环化反应后,本发明优选还包括后处理,所述后处理优选包括:将所得环化反应液冷却至室温,浓缩,加入饱和碳酸氢钠溶液淬灭反应,有机溶剂萃取,将所得有机相进行干燥剂干燥,浓缩后柱层析分离,得到化合物6。本发明对于所述冷却没有特殊限定,采用本领域技术人员熟知的冷却方式即可,具体如自然冷却。本发明对于两次浓缩没有特殊限定,采用本领域技术人员熟知的浓缩方式即可,具体如减压蒸馏,本发明对于所述减压蒸馏没有特殊限定,能够将溶剂除去即可。在本发明中,所述萃取用有机溶剂优选包括二氯甲烷。在本发明中,所述干燥剂优选包括无水硫酸钠。在本发明中,所述柱层析分离优选利用二氧化硅柱层析硅胶柱进
行梯度洗脱,洗脱剂优选包括乙酸乙酯-石油醚混合溶剂,所述乙酸乙酯-石油醚混合溶剂中乙酸乙酯与石油醚的体积比优选为1:0.1~2。
[0072]
得到化合物6后,本发明将所述化合物6、碱性试剂和醇类溶剂混合进行水解反应,得到化合物iii。在本发明中,所述碱性试剂优选包括氢氧化物,更优选包括氢氧化钠和/或氢氧化锂。在本发明中,所述化合物6和碱性试剂的摩尔比优选为1:1~10,更优选为1:2~4。在本发明中,所述醇类溶剂优选为低级醇,更优选为甲醇和/或乙醇;本发明对于所述醇类溶剂的用量没有特殊限定,能够使得水解反应顺利进行即可。本发明对于所述混合没有特殊限定,能够将原料混合均匀即可,具体如搅拌混合。在本发明中,所述水解反应的温度优选为0~80℃,更优选为10~60℃,进一步优选为20~50℃;所述水解反应的时间优选为6~72h,更优选为10~60h,进一步优选为30~50h。完成水解反应后,本发明优选还包括后处理,所述后处理优选包括:将所得水解反应液浓缩,加入水,调节ph值至5~8,固液分离,将所得固体产物进行干燥,得到化合物iii。本发明对于所述浓缩没有特殊限定,采用本领域技术人员熟知的浓缩方式即可,具体如减压蒸馏,本发明对于所述减压蒸馏没有特殊限定,能够将溶剂除去即可。在本发明中,所述调节ph值用ph值调节剂优选包括硫酸、盐酸、醋酸或苹果酸。本发明对于所述固液分离没有特殊限定,采用本领域技术人员熟知的固液分离方式即可,具体如离心分离、过滤或抽滤。在本发明中,所述干燥的温度优选为50~100℃,更优选为60~70℃,本发明对于所述干燥的时间没有特殊限定,干燥至恒重即可。
[0073]
得到化合物ii和化合物iii后,本发明将化合物ii、化合物iii、缩合试剂、活化剂和有机溶剂混合,进行缩合反应,得到所述三唑并吡啶酰胺类化合物。在本发明中,所述缩合剂优选包括1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edci)、2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu)和o-苯并三氮唑-四甲基脲六氟磷酸酯(hbtu)中的一种或几种。在本发明中,所述活化剂优选包括1-羟基-苯并三唑(hobt)和/或1-羟基-7-氮杂苯并三唑(hoat)。在本发明中,所述化合物iii、化合物ii、缩合试剂和活化剂的摩尔比优选为1:0.8~2:1~3:0.2~2,更优选为1:0.8~1.2:1.5~2.5:0.5~1.5。在本发明中,所述有机溶剂优选包括卤代烷类溶剂、杂环类溶剂和苯类溶剂中的一种或几种,更优选包括二氯甲烷、四氢呋喃、二氧六环、甲苯和氯苯中的一种或几种,所述有机溶剂优选为无水溶剂;本发明对于所述有机溶剂的用量没有特殊限定,能够使得缩合反应顺利进行即可。在本发明的具体实施例中,所述混合优选为将化合物iii溶解于有机溶剂中,在冰浴(0℃)条件下加入缩合剂混合12~96h(更优选为20~80h,进一步优选为30~60h),之后加入活化剂和化合物ii混合。在本发明中,所述缩合反应的温度优选为0~40℃,更优选为10~30℃,更优选为20~25℃;所述缩合反应的时间优选为12~96h,更优选为20~80h,进一步优选为30~60h。完成缩合反应后,本发明优选还包括后处理,所述后处理优选包括:在所得缩合反应液中加入淬灭剂淬灭反应,水洗,浓缩后柱层析分离,得到所述三唑并吡啶酰胺类化合物。在本发明中,所述淬灭剂优选包括碱性水溶液,更优选包括nh4cl溶液、nahco3溶液和na2co3溶液中的一种或几种,所述碱性水溶液的质量浓度优选10~50%,更优选为20~40%;所述化合物iii与淬灭剂的用量比优选为1mmol:2~10ml,更优选为1mmol:5~8ml。本发明对于所述水洗没有特殊限定,水洗至中性即可。本发明对于所述浓缩没有特殊限定,采用本领域技术人员熟知的浓缩方式即可,具体如减压蒸馏,优选在旋转蒸发仪进行减压浓缩;本发明对于所述减压蒸馏没有特殊限定,能够将溶剂除去即可。在本发明中,所述柱层析分离优选
利用二氧化硅柱层析硅胶柱进行梯度洗脱,洗脱剂优选包括乙酸乙酯-石油醚混合溶剂,所述乙酸乙酯-石油醚混合溶剂中乙酸乙酯与石油醚的体积比优选为1:1~15。
[0074]
本发明提供了上述技术方案所述的三唑并吡啶酰胺类化合物或上述技术方案所述制备方法得到的三唑并吡啶酰胺类化合物在制备抗植物病原真菌药物中的应用。在本发明中,所述植物病原真菌包括水稻纹枯病菌(rhizoctonia solani),小麦纹枯病菌(rhizoctonia cerealis),油菜菌核病菌(sclerotinia scleotiorum),小麦赤霉病菌(fusarium graminearum),小麦全蚀病菌(gaeumanomyce graminis),番茄灰霉病菌(botrytis cinerea),马铃薯晚疫病菌(phytophthora infestans),辣椒疫霉病菌(phytophthora capsici),番茄早疫病菌(alternaria solani),水稻恶苗病菌(fusarium fujikuroi),马铃薯干腐病菌(fusarium sulphureum),黄瓜炭疽病菌(colletotrichum lagenarium),水稻稻瘟病菌(pyricularia oryzac)中的一种或几种。
[0075]
本发明提供了一种抗植物病原真菌药物,包括活性组分和药用辅料;所述活性组分包括上述技术方案所述的三唑并吡啶酰胺类化合物或上述技术方案所述制备方法得到的三唑并吡啶酰胺类化合物。在本发明中,当所述抗植物病原真菌药物为液态时,所述抗植物病原真菌药物中活性组分的含量优选为0.01~100μmol/l,更优选为1~50μmol/l。在本发明中,所述药用辅料优选包括吐温80、十二烷基硫酸钠硬脂酸,十二烷基苯磺酸钠,季铵化物,卵磷脂,烷基葡糖苷(apg),脂肪酸甘油酯,脂肪酸山梨坦和聚山梨酯中的一种或几种。
[0076]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所得到的所有其他实施例,都属于本发明保护的范围。
[0077]
实施例1
[0078]
(r)-n-(2-(4-乙基-4,5-二氢噁唑-2-基)苯基)-[1,2,4]三唑并[4,3-a]吡啶-8-甲酰胺(i-1)的合成。
[0079][0080]
将邻氨基苯腈(1181mg,10mmol)和(r)-2-氨基-1-丁醇(1070g,12mmol)溶于15ml无水氯苯中,加入无水氯化锌(682mg,5mmol)混合均匀,回流反应36h,减压下蒸除溶剂,加入浓度为15wt%的氢氧化钠溶液(15ml)淬灭反应,用二氯甲烷萃取3次(单次萃取用体积为15ml,记为15ml
×
3),合并有机相后无水硫酸钠干燥,硅胶柱层析分离(200~300目,石油醚:乙酸乙酯体积比=10:1),得到(r)-2-(4-乙基-4,5-二氢-2-噁唑基)苯胺(淡黄色液体,1310mg,产率为69%)。
[0081][0082]
将2-氯烟酸乙酯(1856mg,10mmol)溶于二氧六环(20ml)中,加入(12mmol)水合肼(81wt%),在60℃下搅拌反应12h,冷却至室温后减压蒸出溶剂,硅胶柱层析分离(200~300目,石油醚:乙酸乙酯体积比=5:1)分离得到2-肼基烟酸乙酯(淡黄色固体,740mg,产率为41%)。1hnmr(400mhz,chloroform-d)δ8.67(s,1h,1h innh),8.23(dd,j1=4.8hz,j2=1.9hz,1h,aromatic h),8.05(dd,j1=7.7hz,j2=1.9hz,1h,aromatic h),6.52(dd,j1=7.7hz,j2=4.8hz,1h,aromatic h),4.31-4.25(m,2h,2h in ch2),3.89(s,2h,2h innh2),1.29(t,j=7.1hz,3h,3h in ch3).
13
cnmr(101mhz,chloroform-d)δ166.7,159.9,153.0,140.1,112.0,106.5,61.0,14.3.
[0083]
将2-肼基烟酸乙酯(1810mg,10mmol)溶于原甲酸三甲酯(15ml),在60℃下搅拌反应12h。冷却至室温后减压蒸出溶剂,硅胶柱层析(200~300目,石油醚:乙酸乙酯体积比=1:2)分离得到[1,2,4]三唑基[4,3-a]吡啶-8-羧酸乙酯(白色固体,1280mg,产率为67%)。1hnmr(400mhz,chloroform-d)δ8.98(s,1h,heterocyclic h in triazole ring),8.47(dd,j1=6.8hz,j2=1.1hz,1h,aromatic h),8.02(dd,j1=7.0hz,j2=1.2hz,1h,aromatic h),6.94(t,j=6.9hz,1h,aromatic h),4.54-4.48(m,2h,2h in ch2),1.40(t,j=7.1hz,3h,3h in ch3).
13
cnmr(101mhz,chloroform-d)δ163.4,146.8,136.4,132.7,127.8,119.7,113.3,62.3,14.4.
[0084]
将化合物6(1910mg,10mmol)和氢氧化钠(1200mg,30mmol)溶于甲醇(20ml),在室温下搅拌反应24h,减压下蒸出甲醇,加入水,用浓度为0.1mol/l的盐酸调节溶液ph至6,过滤,将所得固体产物在60℃条件下干燥至恒重,得到[1,2,4]三唑并[4,3-a]吡啶-8-羧酸,直接用于下一步反应。
[0085]
将[1,2,4]三唑并[4,3-a]吡啶-8-羧酸(163mg,1mmol)溶于无水二氯甲烷(5ml),在冰浴条件下加入缩合剂1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edci,211mg,1.1mmol),搅拌10min,加入1-羟基苯并三唑(hobt,27mg,0.2mmol),再加入邻(4,5-二氢-2-噁唑-基)苯胺(190mg,1mmol),在室温下搅拌反应24h,饱和氯化铵溶液淬灭反应,二氯甲烷萃取(5ml
×
2),减压蒸出溶剂,硅胶柱层析分离(200~300目,石油醚:乙酸乙酯体积比=1:2),得到(r)-n-(2-(4-乙基-4,5-二氢噁唑-2-基)苯基)-[1,2,4]三唑并[4,3-a]吡啶-8-甲酰胺(i-1,白色固体,120mg,产率为36%)。1hnmr(500mhz,chloroform-d)δ13.10(s,1h,1h in conh),8.96(s,1h,heterocyclic h in triazole ring),8.74(dd,j1=8.2hz,j2=1.1hz,1h,aromatic h),8.30(dd,j1=6.8hz,j2=1.2hz,1h,aromatic h),8.15(dd,j1=7.0hz,j2=1.1hz,1h,aromatic h),7.86(dd,j1=7.9hz,j2=1.7hz,1h,aromatic h),7.52-7.49(m,1h,aromatic h),7.18-7.14(m,1h,aromatic h),7.02(t,j=6.9hz,1h,aromatic h),4.47(dd,j1=9.6hz,j2=8.0hz,1h,1h inn-ch),4.30-4.24(m,1h,1h in och2),3.99(t,j=8.1hz,1h,1h in och2),1.68-1.49(m,2h,2hin ch2),0.75(t,j=7.4hz,3h,3h in ch3).
13
cnmr(126mhz,chloroform-d)δ162.5,161.2,146.9,138.7,136.0,132.0,
130.9,129.5,126.0,123.7,123.5,122.1,116.5,114.1,71.5,68.3,28.5,10.1.
[0086]
实施例2
[0087]
n-(2-(4-乙基-4,5-二氢噻唑-2-基)苯基)-[1,2,4]三唑基[4,3-a]吡啶-8-甲酰胺(i-2)的合成。
[0088][0089]
将实施例1制备的(r)-2-(4-乙基-4,5-二氢-2-噁唑基)苯胺(1910mg,10mmol)溶于15ml无水甲苯,加入p2s5(4444mg,20mmol),在90℃下搅拌反应24h,冷却至室温后加入浓度为20wt%的naoh溶液(15ml)淬灭反应,过滤除去滤渣,滤液用分液漏斗分离,将所得有机相无水硫酸钠干燥,减压蒸出溶剂,硅胶柱层析分离,(200~300目,石油醚:乙酸乙酯体积比=10:1),得到2-(4-乙基-4,5-二氢噻唑-2-基)苯胺(0.227g,黄色油状,产率为11%)。1hnmr:(500mhz,chloroform-d)δ7.47-7.45(m,1h,aromatic h),7.20-7.17(m,1h,aromatic h),6.76-6.66(m,1h,aromatic h),4.67-4.58(m,1h,1h in n-ch),3.39-3.35(m,1h,1h in sch2),2.97-2.94(m,1h in sch2),1.93-1.84(m,1h,1h in ch2),1.79-1.70(m,1h,1h in ch2),1.09(t,j=7.4hz,3h,3h in ch3).
13
cnmr(126mhz,chloroform-d)δ168.0,147.0,132.4,131.68,116.8,116.5,115.3,79.3,36.1,28.5,11.4.
[0090]
将实施例1制备的[1,2,4]三唑并[4,3-a]吡啶-8-羧酸(163mg,1mmol)溶于无水二氯甲烷(5ml),在冰浴条件下加入缩合剂1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edci,211mg,1.1mmol),搅拌10min,随后加入1-羟基苯并三唑(hobt,27mg,0.2mmol),再加入邻(4,5-二氢-2-噻唑-基)苯胺(206mg,1mmol),在室温下搅拌反应,饱和氯化铵溶液淬灭反应,用二氯甲烷萃取2次(5ml
×
2),减压蒸出溶剂,硅胶柱层析分离(200~300目,石油醚:乙酸乙酯体积比=1:2)得到n-(2-(4-乙基-4,5-二氢噻唑-2-基)苯基)-[1,2,4]三唑基[4,3-a]吡啶-8-甲酰胺(i-2,白色固体,200mg,产率为57%)。1hnmr(500mhz,chloroform-d)δ13.11(s,1h,1h in conh),8.95(s,1h,heterocyclic h in triazole ring),8.72(dd,j1=8.6hz,j2=1.1hz,1h,aromatic h),8.29(dd,j1=6.9hz,j2=1.1hz,1h,aromatic h),8.15(dd,j1=6.9hz,j2=1.2hz,1h,ar omatic h),7.63(dd,j1=7.8hz,j2=1.6hz,1h,aromatic h),7.48-7.44(m,1h,aromatic h),7.17-7.14(m,1h,aromatic h),7.00(t,j=6.9hz,1h,aromatic h),4.74-4.67(m,1h,1h in n-ch),3.43(dd,j1=10.5hz,j2=8.4hz,1h,1h in sch2),2.99(dd,j1=10.7hz,j2=9.0hz,1h,1h in sch2),1.76-1.68(m,1h,1h in ch2),1.62-1.54(m,1h,1h in ch2),0.69(t,j=7.4hz,3h,3h in ch3).
13
cnmr(126mhz,chloroform-d)δ166.2,161.2,146.8,137.4,136.1,131.3,131.1,126.0,124.0,123.2,122.3,122.2,114.2,79.9,37.0,28.0,10.7.
[0091]
实施例3
[0092]
(r)-n-(2-(4-乙基-4,5-二氢噁唑-2-基)苯基)-3-苯基-[1,2,4]三唑基[4,3-a]
吡啶-8-甲酰胺(i-3)的合成。
[0093][0094]
将2-肼基烟酸乙酯(1810mg,10mmol)、苯甲亚胺酸乙酯盐酸盐(1860mg,10mmol)、乙酸钠(1230mg,15mmol)和乙酸(300mg,5mmol)溶于无水乙醇(25ml),在50℃下搅拌反应24h,冷却至室温后减压蒸出溶剂,加入饱和碳酸氢钠溶液,用二氯甲烷萃取(5ml
×
2),用分液漏斗分离,无水硫酸钠干燥,减压蒸出溶剂,硅胶柱层析(200~300目,石油醚:乙酸乙酯体积比=1:1)分离得到3-苯基-[1,2,4]三唑基[4,3-a]吡啶-8-羧酸乙酯(黄色固体,1095mg,产率为41%。1hnmr(500mhz,chloroform-d)δ8.41(dd,j=7.0hz,1.1hz,1h,aromatic h),8.05(dd,j=6.9hz,1.2hz,1h,aromatic h),7.79-7.75(m,2h,aromatic h),7.60-7.52(m,3h,aromatic h),6.95(t,j=6.9hz,1h,aromatic h),4.55(q,j=7.1hz,2h,2h in ch2),1.47(t,j=7.1hz,3h,3h in ch3).
13
cnmr(126mhz,chloroform-d)δ163.6,147.9,147.4,132.1,130.6,129.5(2c),128.7(2c),126.6,126.2,120.2,113.4,62.3,14.4
[0095]
将3-苯基-[1,2,4]三唑基[4,3-a]吡啶-8-羧酸乙酯(2660mg,10mmol)和氢氧化钠(1200mg,30mmol)溶于甲醇(20ml),室温下搅拌过夜,减压下蒸出甲醇,加入水,利用0.1mol/l盐酸调节溶液ph至6,过滤,将所得固体产物在60℃条件下干燥至恒重,得到3-苯基-[1,2,4]三唑基[4,3-a]吡啶-8-羧酸。
[0096]
将3-苯基-[1,2,4]三唑基[4,3-a]吡啶-8-羧酸(239mg,1mmol)溶于无水二氯甲烷(5ml),在冰浴条件下加入缩合剂1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edci,211mg,1.1mmol),搅拌10min,随后加入1-羟基苯并三唑(hobt,27mg,0.2mmol),再加入实施例1制备的(r)-2-(4-乙基-4,5-二氢-2-噁唑基)苯胺(190mg,1mmol),室温下搅拌反应24h,饱和氯化铵溶液淬灭反应,用二氯甲烷萃取(5ml
×
2),减压蒸出溶剂,硅胶柱层析分离(200~300目,石油醚:乙酸乙酯体积比=1:2),得到(r)-n-(2-(4-乙基-4,5-二氢噁唑-2-基)苯基)-3-苯基-[1,2,4]三唑基[4,3-a]吡啶-8-甲酰胺(i-3,白色固体,120mg,产率为26%)。1hnmr(500mhz,chloroform-d)δ13.10(s,1h,1h in conh),8.76(dd,j1=8.6hz,j2=1.1hz,1h,aromatic h),8.37(dd,j1=7.0hz,j2=1.1hz,1h,aromatic h),8.16(dd,j1=6.9hz,j2=1.1hz,1h,aromatic h),7.87(dd,j1=7.9hz,j2=1.7hz,1h,aromatic h),7.85-7.77(m,2h,aromatic h),7.65-7.54(m,3h,aromatic h),7.53-7.49(m,1h,aromatic h),7.18-7.14(m,1h,aromatic h),7.01(t,j=6.9hz,1h,aromatic h),4.50(dd,j1=9.6,j2=8.0hz,1h,1h in n-ch),4.39-4.25(m,1h,1h in och2),4.01(t,j=8.2hz,1h,1h in och2),1.71-1.52(m,2h,2h in ch2),0.76(t,j=7.4hz,3h,3h in ch3).
13
cnmr(126mhz,chloroform-d)δ162.5,161.3,147.9,147.3,138.7,131.9,130.7,130.5,129.6(2c),128.6(2c),126.3,125.2,123.7,122.1,116.7,114.1,71.6,68.3,28.5,10.1.
[0097]
实施例4
[0098]
n-(2-(4-乙基-4,5-二氢噻唑-2-基)苯基)-3-苯基-[1,2,4]三唑基[4,3-a]吡啶-8-甲酰胺(i-4)的合成。
[0099][0100]
将实施例3制备的3-苯基-[1,2,4]三唑基[4,3-a]吡啶-8-羧酸(239mg,1mmol)溶于无水二氯甲烷(5ml),冰浴条件下加入缩合剂1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edci,211mg,1.1mmol),搅拌10min,随后加入1-羟基苯并三唑(hobt,27mg,0.2mmol),再加入实施例2中的2-(4-乙基-4,5-二氢噻唑-2-基)苯胺(206mg,1mmol),室温下搅拌反应24h,饱和氯化铵溶液淬灭反应,用二氯甲烷萃取(5ml
×
2),减压蒸出溶剂,硅胶柱层析分离(200~300目,石油醚:乙酸乙酯体积比=1:2),得到n-(2-(4-乙基-4,5-二氢噻唑-2-基)苯基)-3-苯基-[1,2,4]三唑基[4,3-a]吡啶-8-甲酰胺(i-4,白色固体,137mg,产率为32%)。1hnmr(400mhz,chloroform-d)δ13.18(s,1h,1h in conh),8.83-8.74(m,1h,aromatic h),8.39(dd,j1=7.0hz,j2=1.0hz,1h,aromatic h),8.19(dd,j1=6.9hz,j2=1.0hz,1h,aromatic h),7.83(dd,j1=7.8hz,j2=1.7hz,2h,aromatic h),7.71-7.55(m,4h,aromatic h),7.51-7.47(m,1h,aromatic h),7.21-7.16(m,1h,aromatic h),7.03(t,j1=7.0hz,1h,aromatic h),4.80-4.76(m,1h,1h in n-ch),3.49(dd,j1=10.7hz,j2=8.5hz,1h,1h in sch2),3.03(dd,j1=10.6hz,j2=9.2hz,1h,1h in sch2),1.81-1.72(m,1h,1h in ch2),1.69-1.60(m,1h,1h in ch2),0.73(t,j=7.4hz,3h,3h in ch3).3cnmr(101mhz,chloroform-d)δ165.9,161.2,147.9,147.2,137.4,131.2,131.0,130.6,130.6,129.5(2c),128.5(2c),126.3,125.1,123.8,123.5,122.3,122.2,114.1,79.9,37.0,28.1,10.7.
[0101]
实施例5
[0102]
(s)-n-(2-(4-(叔丁基)-4,5-二氢噁唑-2-基)苯基)-3-苯基-[1,2,4]三唑并[4,3-a]吡啶-8-甲酰胺(i-5)的合成。
[0103][0104]
将邻氨基苯腈(118mg,1mmol)和l-叔亮氨醇(185mg,1.2mmol)溶于1ml无水氯苯
中,加入无水氯化锌(69mg,0.5mmol),回流反应36h,减压下蒸除溶剂,加入浓度为15wt%的氢氧化钠溶液(15ml)淬灭反应,用二氯甲烷萃取(15ml
×
3),合并有机相,无水硫酸钠干燥,硅胶柱层析分离(200~300目,石油醚:乙酸乙酯体积比=10:1),得到(s)-2-(4-(叔丁基)-4,5-二氢噁唑-2-基)苯胺(白色固体,120mg,产率为55%)。
[0105]
将实施例3制备的3-苯基-[1,2,4]三唑基[4,3-a]吡啶-8-羧酸(239mg,1mmol)溶于无水二氯甲烷(5ml),在冰浴条件下加入缩合剂1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edci,211mg,1.1mmol),搅拌10min,随后加入1-羟基苯并三唑(hobt,27mg,0.2mmol),再加入(s)-2-(4-(叔丁基)-4,5-二氢噁唑-2-基)苯胺(218mg,1mmol),在室温下搅拌反应24h,饱和氯化铵溶液淬灭反应,用二氯甲烷萃取(5ml
×
2),减压蒸出溶剂,硅胶柱层分离析(200~300目,石油醚:乙酸乙酯体积比=1:2),得到(s)-n-(2-(4-(叔丁基)-4,5-二氢噁唑-2-基)苯基)-3-苯基-[1,2,4]三唑并[4,3-a]吡啶-8-甲酰胺(i-5,,白色固体,272mg,产率为62%)。1hnmr(500mhz,chloroform-d)δ13.25(s,1h,1h in conh),8.74(dd,j1=8.5hz,j2=1.2hz,1h,aromatic h),8.36(dd,j1=6.9hz,j2=1.3hz,1h,aromatic h),8.08(dd,j1=6.9hz,j2=1.2hz,1h,aromatic h),7.87(dd,j1=7.8hz,j2=1.7hz,1h,aromatic h),7.79(dd,j1=7.9hz,j2=1.7hz,2h,aromatic h),7.63-7.54(m,3h,aromatic h),7.53-7.49(m,1h,aromatic h),7.17-7.14(m,1h,aromatic h),7.00(t,j=7.0hz,1h,aromatic h),4.39-4.28(m,1h,1h in n-ch),4.17-4.06(m,2h,2h in och2),0.64(s,9h,9h in c(ch3)3).
13
cnmr(126mhz,chloroform-d)δ162.5,161.5,147.9,147.3,138.9,132.0,130.6,123.0,129.6(2c),129.5,128.6(2c),126.4,125.1,124.2,123.6,122.0,116.2,114.1,76.1,67.9,33.7,25.6.
[0106]
实施例6
[0107]
(s)-3-苯基-n-(2-(4-苯基-4,5-二氢噁唑-2-基)苯基)-[1,2,4]三唑基[4,3-a]吡啶-8-甲酰胺(i-6)的合成。
[0108][0109]
将邻氨基苯腈(1180mg,1mmol)和l-苯甘氨醇(164mg,1.2mmol)溶于1ml无水氯苯中,加入无水氯化锌(69mg,0.5mmol),回流反应36h,减压下蒸除溶剂,加入浓度为15wt%的氢氧化钠溶液(15ml)淬灭反应,用二氯甲烷萃取(15ml
×
3),合并有机相,无水硫酸钠干燥,硅胶柱层析(200~300目,石油醚:乙酸乙酯体积比=10:1)分离,得到(s)-2-(4-苯基-4,5-二氢噁唑-2-基)苯胺(白色固体,162mg,产率为68%)。
[0110]
将实施例3制备的3-苯基-[1,2,4]三唑基[4,3-a]吡啶-8-羧酸(239mg,1mmol)溶于无水二氯甲烷(5ml),冰浴条件下加入缩合剂1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edci,211mg,1.1mmol),搅拌10min,随后加入1-羟基苯并三唑(hobt,27mg,0.2mmol),再加入(s)-2-(4-苯基-4,5-二氢噁唑-2-基)苯胺(238mg,1mmol),室温下搅拌反应24h,饱和
and 1h in och2),3.96(t,j=7.3hz,1h,1h in och2),1.35(d,j=6.3hz,3h,3h in ch3).
13
cnmr(101mhz,chloroform-d)δ162.2,161.1,147.8,147.3,138.6,131.8,130.7,130.6,129.5,129.5(2c),128.6(2c),126.2,125.2,123.6,123.3,122.0,116.8,114.08,73.4,62.3,21.3.
[0116]
实施例8
[0117]
(s)-n-(2-(4-乙基-4,5-二氢噁唑-2-基)苯基)-3-苯基-[1,2,4]三唑基[4,3-a]吡啶-8-甲酰胺i-8的合成。
[0118][0119]
将邻氨基苯腈(1180mg,1mmol)和(s)-2-氨基-1-丁醇(1070mg,1.2mmol)溶于1ml无水氯苯中,加入无水氯化锌(69mg,0.5mmol),回流反应36h,减压下蒸除溶剂,加入浓度为15wt%的氢氧化钠溶液(15ml)淬灭反应,用二氯甲烷萃取(15ml
×
3),合并有机相,无水硫酸钠干燥,硅胶柱层析分离(200~300目,石油醚:乙酸乙酯体积比=10:1),得到(s)-2-(4-乙基-4,5-二氢噁唑-2-基)苯胺(黄色液体,144mg,产率为76%。
[0120]
将实施例3制备的3-苯基-[1,2,4]三唑基[4,3-a]吡啶-8-羧酸(239mg,1mmol)溶于无水二氯甲烷(5ml),冰浴条件下加入缩合剂1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edci,211mg,1.1mmol),搅拌10min,随后加入1-羟基苯并三唑(hobt,27mg,0.2mmol),再加入(s)-2-(4-乙基-4,5-二氢噁唑-2-基)苯胺(238mg,1mmol),室温下搅拌反应24h,饱和氯化铵溶液淬灭反应,用二氯甲烷萃取(5ml
×
2),减压蒸出溶剂,硅胶柱层析分离(200~300目,石油醚:乙酸乙酯体积比=1:2),得到(s)-n-(2-(4-乙基-4,5-二氢噁唑-2-基)苯基)-3-苯基-[1,2,4]三唑基[4,3-a]吡啶-8-甲酰胺(i-8,白色固体,1150mg,产率为28%)。1hnmr(500mhz,chloroform-d)δ13.11(s,1h,1h in conh),8.77(dd,j1=8.4hz,j2=1.1hz,1h,aromatic h),8.38(dd,j1=6.9hz,j2=1.2hz,1h,aromatic h),8.16(dd,j1=6.9hz,j2=1.2hz,1h,aromatic h),7.88(dd,j1=7.8hz,j2=1.7hz,1h,aromatic h),7.85-7.77(m,2h,aromatic h),7.64-7.57(m,3h,aromatic h),7.53-7.50(m,1h,aromatic h),7.18-7.15(m,1h,aromatic h),7.02(t,j=6.9hz,1h,aromatic h),4.50(dd,j1=9.6hz,j2=8.1hz,1h,1h inn-ch),4.35-4.28(m,1h,1h in och2),4.01(t,j=8.1hz,1h,1h in och2),1.69-1.54(m,2h,2h in ch2),0.76(t,j=7.4hz,3h,3h in och2).
13
cnmr(126mhz,chloroform-d)δ162.5,161.3,147.9,147.3,138.7,131.9,130.7,130.5,129.6(2c),128.7(2c),126.4,125.2,123.7,122.1,116.7,114.1,71.6,68.3,28.5,10.1.
[0121]
测试例
[0122]
三唑并吡啶酰胺类化合物i-1~i-8的抑菌活性测定
[0123]
采用抑制菌丝生长速率法进行离体抑菌活性评价,选取测试菌株于pda平板进行
活化,包括水稻纹枯病菌(rhizoctonia solani),小麦纹枯病菌(rhizoctonia cerealis),油菜菌核病菌(sclerotinia scleotiorum),小麦赤霉病菌(fusarium graminearum),小麦全蚀病菌(gaeumanomyce graminis),番茄灰霉病菌(botrytis cinerea),马铃薯晚疫病菌(phytophthora infestans),辣椒疫霉病菌(phytophthora capsici),番茄早疫病菌(alternaria solani),水稻恶苗病菌(fusarium fujikuroi),马铃薯干腐病菌(fusarium sulphureum),黄瓜炭疽病菌(colletotrichum lagenarium),水稻稻瘟病菌(pyricularia oryzac)。将化合物i-1~i-8分别配置成系列梯度浓度(浓度分别为30μm,10μm、3.3μm、1.1μm、0.37μm、0.12μm和0.04μm,其中,μm表示μmol/l)的pda含药平板,将测试菌株制成5mm直径菌饼置于含药培养皿中央,在25℃恒温培养至空白对照皿的测试菌株长至接近培养皿边缘时,用十字交叉法测量各含药平板的菌落直径,计算化合物对菌丝生长的抑制率,对病害的抑制率按照式(1)计算:
[0124]
病害抑制率(%)=(对照组的菌落直径-药物组的菌落直径)/对照组的菌落直径
×
100%式(1)。
[0125]
使用统计软件spss 20.0计算抑制率为50%时化合物i-1~i-8的浓度,即ec
50
值,重复3次取平均值。实验中以啶酰菌胺为阳性对照。三唑并吡啶酰胺类化合物i-1~i-8的抑菌活性测试结果如表1~2所示。
[0126]
表1三唑并吡啶酰胺类化合物对六种农用真菌在30μmol/l浓度下的抑制率
[0127][0128][0129]
表2三唑并吡啶酰胺类化合物对六种农用真菌的抑制中浓度
[0130][0131]
从表1和表2可以看出,三唑并吡啶酰胺类化合物对常见的农业真菌有较好的抑制作用,且三唑环上的取代基以及酰胺的胺片段对抑菌活性有显著影响。三唑并吡啶类化合物中三唑环上无取代基时抑菌活性显著降低,苯基的引入可以显著提高抑菌活性。胺片段引入体积较大的手性取代基时抑菌活性显著降低,引入体积较小的手性取代基时可以显著提高抑菌活性。胺片段引入噻唑啉环可以显著提高抑菌活性。
[0132]
经过结构优化,当三唑并吡啶酰胺类化合物的三唑环上为苯基取代,胺片段为r构型乙基取代的苯基噻唑啉时,对水稻纹枯病菌,油菜菌核病菌,番茄灰霉病菌,小麦赤霉病菌,辣椒疫霉病菌,稻瘟病菌的抑制率分别为0.20μmol/l、1.45μmol/l、0.18μmol/l、3.13μmol/l和0.60μmol/l。该化合物对小麦赤霉病菌和辣椒疫霉病菌的活性明显高于阳性对照啶酰菌胺。三唑并吡啶酰胺类化合物有望作为一类新型的杀菌剂候选化合物或者直接作为杀菌剂使用,对新农药创制将有重要意义。
[0133]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1