一种vulgarisin型二萜化合物、提取方法及用途
1.本发明涉及天然药物技术领域,特别是涉及一种vulgarisin型二萜化合物、提取方法及用途。
背景技术:
2.vulgarisin型二萜化合物是一类具有药效活性的药物。本技术发现从夏枯草中也可提取得到vulgarisin型二萜化合物,得到的vulgarisin型二萜化合物属于全新的化合物,具有其相应的药用价值。
技术实现要素:
3.鉴于以上所述现有技术的缺点,本发明的目的在于提供一种vulgarisin型二萜化合物;同时,本发明还将提供一种从夏枯草中提取vulgarisin型二萜化合物的方法;此外,本发明还将提供上述vulgarisin型二萜化合物在制备具有神经元保护活性药物上的应用。
4.为实现上述目的及其他相关目的,
5.本发明的第一方面,提供一种vulgarisin型二萜化合物,所述vulgarisin型二萜化合物的的结构通式如i所示:
[0006][0007]
所述r1、r5、r7彼此独立地选自h或烷基;
[0008]
所述r2、r3、r4彼此独立的选自羟基、中的一种;
[0009]
所述r6为h或羟基。
[0010]
于本发明的一实施例中,所述r1、r5、r7彼此独立地选自碳原子数为1~6的烷基。
[0011]
于本发明的一实施例中,所述r1、r7均为碳原子数为1~3的直链烷基,所述r5为碳原子数为3~5的支链烷基;。
[0012]
于本发明的一实施例中,所述r1、r7均为甲基,所述r5为异丙基,所述r6为羟基。
[0013]
于本发明的一实施例中,所述r2或r3为羟基。
[0014]
于本发明的一实施例中,所述r2为羟基,所述r3为
中的一种。
[0015]
于本发明的一实施例中,所述r3为羟基,所述r2为中的一种。
[0016]
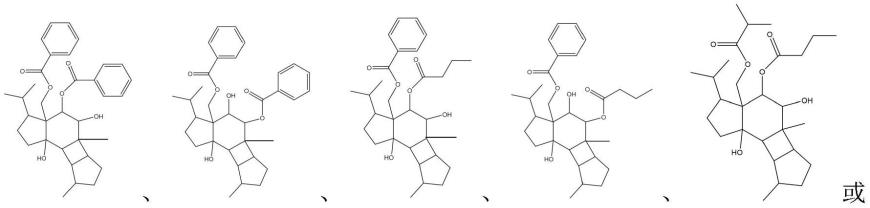
于本发明的一实施例中,所述vulgarisin型二萜化合物为于本发明的一实施例中,所述vulgarisin型二萜化合物为
[0017]
本发明的第二方面,提供一种从夏枯草中提取vulgarisin型二萜化合物的方法,包括如下步骤:
[0018]
将夏枯草进醇提得到浸膏;
[0019]
将浸膏采用石油醚萃取得到预提物;
[0020]
将预提物采用石油醚-乙酸乙酯体系按照石油醚占比逐渐减小进行梯度洗脱,得到vulgarisin型二萜化合物粗提物;
[0021]
vulgarisin型二萜化合物粗提物采用甲醇-水体系按照甲醇占比逐渐增大进行洗脱,即得vulgarisin型二萜化合物。
[0022]
于本发明的一实施例中,预提物按照石油醚-乙酸乙酯体积比依次为100:0
→
(85~95):(5~15)
→
(65~75):(25~35)
→
(45~55):(45~55)
→
(15~25):(75~85)
→
0:100进行梯度洗脱,依次得到fr.a、fr.b、fr.c、fr.d、fr.e、fr.f;
[0023]
将fr.d按照甲醇-水体积比依次为(35~45):(55~65)
→
(45~55):(45~55)
→
(55~65):(35~45)
→
(65~75):(25~35)
→
(75~85):(15~25)
→
(85~95):(5~15)
→
100:0进行梯度洗脱,依次得到fr.d1、fr.d2、fr.d3、fr.d4、fr.d5、fr.d6、fr.d7;
[0024]
将fr.d5按照甲醇-水体积比依次为(35~45):(55~65)
→
(55~65):(35~45)
→
(75~85):(15~25)
→
(85~95):(5~15)
→
100:0进行梯度洗脱,依次得到fr.d5-1、fr.d5-2、fr.d5-3、fr.d5-4、fr.d5-5;
[0025]
将fr.d5-1按照甲醇-水体积比为(80~95):(5~20)进行洗脱,即得化合物1、化合物3、化合物7、化合物11;
[0026]
将fr.d5-2按照甲醇-水体积比为(70~90):(10~30)进行洗脱,即得化合物2、化合物8、化合物10;
[0027]
将fr.d5-3按照甲醇-水体积比为(85~95):(5~15)进行洗脱,即得化合物4、化合物5、化合物9、化合物12;
[0028]
将fr.d5-4按照甲醇-水体积比为(90~100):(2~10)进行洗脱,即得化合物6;
[0029]
其中,所述化合物1~12依次为合物1~12依次为
[0030]
本发明的第三方面,提供一种上述vulgarisin型二萜化合物在制备具有神经元保护活性药物上的应用。
[0031]
于本发明的一实施例中,所述具有神经元保护活性药物为用于治疗脑卒中的药物、用于治疗老年痴呆的药物或用于治疗神经退行性疾病的药物。
[0032]
如上所述,本发明的一种vulgarisin型二萜化合物、提取方法及用途,具有以下有益效果:
[0033]
1、本发明首次从夏枯草属植物的全草中获得了vulgarisin型二萜化合物(化合物1~~6)。通过hr-esi-ms、1d和2d nmr的方法确定了它们的平面结构,进一步通过x-ray或圆二色谱法确定了6个化合物的绝对构型。
[0034]
2、本发明从夏枯草中分离得到的化合物1~12,初步神经保护活性筛选发现均具有一定的神经元保护活性,可以用于脑卒中、老年痴呆等脑病的治疗及预防神经保护的作用主要是防治神经元细胞死亡,提高存活率和活力。
具体实施方式
[0035]
以下由特定的具体实施例说明本发明的实施方式,熟悉此技术的人士可由本说明书所揭露的内容轻易地了解本发明的其他优点及功效。
[0036]
化合物1-12的结构如下所示:
[0037][0038]
一、提取、分离实验部分
[0039]
实施例1
[0040]
化合物1~12的提取与分离
[0041]
步骤一、夏枯草全草自然风干后(干重约20kg)粉碎,然后浸泡于60l 95%乙醇中,加热回流提取两次,浓缩提取液得到浸膏3.3kg。
[0042]
步骤二、将浸膏分散于4l水中,依次用等体积的石油醚、乙酸乙酯和水饱和正丁醇萃取。初步实验证明石油醚部位中含有较丰富的vulgarisin型二萜化合物。因此后续的药效物质基础研究主要集中于夏枯草的石油醚部位。
[0043]
步骤三、首先将石油醚段浸膏(417.0g),采用硅胶中压柱层析(200-300目)以石油醚-乙酸乙酯体系(100:0
→
90:10
→
70:30
→
50:50
→
20:80
→
0:100)进行梯度洗脱,得到6个组分(fr.a-fr.f),具体如表格1所示:
[0044]
洗脱体系洗脱后的组分石油醚-乙酸乙酯的体积比为100:0fr.a石油醚-乙酸乙酯的体积比为90:10fr.b
石油醚-乙酸乙酯的体积比为70:30fr.c石油醚-乙酸乙酯的体积比为50:50fr.d石油醚-乙酸乙酯的体积比为20:80fr.e石油醚-乙酸乙酯的体积比为0:100fr.f
[0045]
表1
[0046]
步骤四、结合tlc分析和uplc-esi-q-exactive-ms分析;组分frd(104.0g)经mci柱层析以甲醇-水体系为流动相进行梯度洗脱(40:60
→
50:50
→
60:40
→
70:30
→
80:20
→
90:10
→
纯甲醇,v/v),得到7个组分(frd1-d7),具体如表格2所示:
[0047]
洗脱体系洗脱后的组分甲醇-水的体积比为40:60frd1甲醇-水的体积比为50:50frd2甲醇-水的体积比为60:40frd3甲醇-水的体积比为70:30frd4甲醇-水的体积比为80:20frd5甲醇-水的体积比为90:10frd6甲醇-水的体积比为100:0frd7
[0048]
表2
[0049]
步骤五、组分frd5然后经聚酰胺柱层析以甲醇-水体系为流动相进行梯度洗脱(40:60
→
60:40
→
80:20
→
90:10
→
100:0,v/v),得到5个组分(frd5-1~frd5-5),具体如表3所示:
[0050]
洗脱体系洗脱后的组分甲醇-水的体积比为40:60frd5-1甲醇-水的体积比为60:40frd5-2甲醇-水的体积比为80:20frd5-3甲醇-水的体积比为90:10frd5-4甲醇-水的体积比为100:0frd5-5
[0051]
表3
[0052]
步骤六、组分frd5-1(909.8mg)经半制备hplc以甲醇-水(87:13,v/v;2ml/min)体系等度洗脱分离获得化合物1(rt 18.5min)、化合物3(rt 19.6min)、化合物7(rt 20.3min)和化合物11(rt 23.5min)。
[0053]
步骤七、组分frd5-2(493.0mg)经半制备hplc(苯基柱)以甲醇-水(80:20,v/v;2ml/min)体系等度洗脱分离获得化合物2(rt 25.5min)、化合物8(rt 27.3min)和化合物10(rt 20.2min)。
[0054]
步骤八、组分frd5-3(726.3mg)先经葡聚糖凝胶lh-20纯化然后经半制备hplc以甲醇-水(92:8,v/v;2ml/min)体系等度洗脱分离获得化合物4(rt 18.5min)、化合物5(rt16.6min)、化合物9(rt 15.0min)和化合物12(rt 19.9min)。
[0055]
步骤九、组分frd5-4(87.0mg)经半制备hplc以甲醇-水(94:6,v/v;2ml/min)体系等度洗脱分离获得化合物6(rt 14.2min)。
[0056]
将步骤六~九中分离得到的化合物产量汇总,如表格4所示:
[0057]
化合物123456产量/mg16.910.018.923.022.518.9化合物789101112产量/mg15.025.0112.1167.0133.0105.0
[0058]
表4
[0059]
图1为化合物1的1hnmr谱图,图2为化合物1的
13
c nmr谱图,图3为化合物1的hsqc谱图,图4为化合物1的hmbc谱图,图5为化合物1的1h-1
h cosy谱图,图6为化合物1的noesy谱图,图7为化合物1的高分辨质谱图。
[0060]
图8为化合物2的1h nmr谱图,图9为化合物2的
13
c nmr谱图,图10为化合物2的hsqc谱图,图11为化合物2的hmbc谱图,图12为化合物2的1h-1
h cosy谱图,图13为化合物2的noesy谱图,图14为化合物2的高分辨质谱图。
[0061]
图15为化合物3的1hnmr谱图,图16为化合物3的
13
c nmr谱图,图17为化合物3的hsqc谱图,图18为化合物3的hmbc谱图,图19为化合物3的1h-1
h cosy谱图,图20为化合物3的noesy谱图,图21为化合物3的高分辨质谱图。
[0062]
图22为化合物4的1hnmr谱图,图23为化合物4的
13
c nmr谱图,图24为化合物4的hsqc谱图,图25为化合物4的hmbc谱图,图26为化合物4的1h-1
h cosy谱图,图27为化合物4的noesy谱图,图28为化合物4的高分辨质谱图。
[0063]
图29为化合物5的1hnmr谱图,图30为化合物5的
13
c nmr谱图,图31为化合物5的hsqc谱图,图32为化合物5的hmbc谱图,图33为化合物5的1h-1
h cosy谱图,图34为化合物5的noesy谱图,图35为化合物5的高分辨质谱图。
[0064]
图36为化合物6的1hnmr谱图,图37为化合物6的
13
c nmr谱图,图38为化合物6的hsqc谱图,图39为化合物6的hmbc谱图,图40为化合物6的1h-1
h cosy谱图,图41为化合物6的noesy谱图,图42为化合物6的高分辨质谱图。
[0065]
图43为化合物1的x-ray单晶结构图,图44为化合物1的圆二色谱测试图,图45为化合物2的圆二色谱测试图,图46为化合物3的圆二色谱测试图,图47为化合物4的圆二色谱测试图,图48为化合物5的圆二色谱测试图,图49为化合物6的圆二色谱测试图。
[0066]
实施例2
[0067]
实施例2与实施例1的区别在于步骤三的洗脱条件不同,具体如表格5所示:
[0068]
洗脱体系洗脱后的组分石油醚-乙酸乙酯的体积比为100:0fr.a石油醚-乙酸乙酯的体积比为90:10fr.b石油醚-乙酸乙酯的体积比为70:30fr.c石油醚-乙酸乙酯的体积比为49:51fr.d石油醚-乙酸乙酯的体积比为20:80fr.e石油醚-乙酸乙酯的体积比为0:100fr.f
[0069]
表5实施例2中化合物的产量如表格6所示:
[0070]
化合物123456产量/mg17.69.216.820.122.513.5
化合物789101112产量/mg16.622.8130.3177.7135.5110.0
[0071]
表6
[0072]
实施例3
[0073]
实施例3与实施例1的区别在于步骤三的洗脱条件不同,具体如表格7所示:
[0074]
洗脱体系洗脱后的组分石油醚-乙酸乙酯的体积比为100:0fr.a石油醚-乙酸乙酯的体积比为90:10fr.b石油醚-乙酸乙酯的体积比为70:30fr.c石油醚-乙酸乙酯的体积比为45:55fr.d石油醚-乙酸乙酯的体积比为20:80fr.e石油醚-乙酸乙酯的体积比为0:100fr.f
[0075]
表7实施例3中化合物的产量如表格8所示:
[0076]
化合物123456产量/mg16.611.418.122.122.217.7化合物789101112产量/mg15.628.9130.0118.8166.6150.5
[0077]
表8
[0078]
实施例4
[0079]
实施例4与实施例1的区别在于步骤三的洗脱条件不同,具体如表格9所示:
[0080]
洗脱体系洗脱后的组分石油醚-乙酸乙酯的体积比为100:0fr.a石油醚-乙酸乙酯的体积比为90:10fr.b石油醚-乙酸乙酯的体积比为70:30fr.c石油醚-乙酸乙酯的体积比为55:45fr.d石油醚-乙酸乙酯的体积比为20:80fr.e石油醚-乙酸乙酯的体积比为0:100fr.f
[0081]
表9实施例4中化合物的产量如表格10所示:
[0082][0083][0084]
表10
[0085]
实施例5
[0086]
实施例5与实施例1的区别在于步骤四的洗脱条件不同,具体如表格11所示:
[0087]
洗脱体系洗脱后的组分
甲醇-水的体积比为40:60frd1甲醇-水的体积比为50:50frd2甲醇-水的体积比为60:40frd3甲醇-水的体积比为70:30frd4甲醇-水的体积比为78:22frd5甲醇-水的体积比为90:10frd6甲醇-水的体积比为100:0frd7
[0088]
表11实施例5中化合物的产量如表格12所示:
[0089]
化合物123456产量/mg18.811.320.524.125.223.2化合物789101112产量/mg13.626.6167.7155.5120.5142.2
[0090]
表12
[0091]
实施例6
[0092]
实施例6与实施例1的区别在于步骤四的洗脱条件不同,具体如表格13所示:
[0093]
洗脱体系洗脱后的组分甲醇-水的体积比为40:60frd1甲醇-水的体积比为50:50frd2甲醇-水的体积比为60:40frd3甲醇-水的体积比为70:30frd4甲醇-水的体积比为82:18frd5甲醇-水的体积比为90:10frd6甲醇-水的体积比为100:0frd7
[0094]
表13实施例6中化合物的产量如表格14所示:
[0095]
化合物123456产量/mg19.611.220.526.620.215.5化合物789101112产量/mg16.826.8144.4200.3166.1115.0
[0096]
表14
[0097]
实施例7
[0098]
实施例7与实施例1的区别在于步骤四的洗脱条件不同,具体如表格15所示:
[0099]
洗脱体系洗脱后的组分甲醇-水的体积比为35:65frd5-1甲醇-水的体积比为55:45frd5-2甲醇-水的体积比为75:25frd5-3甲醇-水的体积比为90:10frd5-4甲醇-水的体积比为100:0frd5-5
[0100]
表15实施例7中化合物的产量如表格16所示:
[0101]
化合物123456产量/mg13.86.518.221.115.517.4化合物789101112产量/mg21.525.6132.3158.8146.2100.1
[0102]
表16
[0103]
实施例8
[0104]
实施例8与实施例1的区别在于步骤四的洗脱条件不同,具体如表格17所示:
[0105]
洗脱体系洗脱后的组分甲醇-水的体积比为42:58frd5-1甲醇-水的体积比为62:38frd5-2甲醇-水的体积比为82:18frd5-3甲醇-水的体积比为90:10frd5-4甲醇-水的体积比为100:0frd5-5
[0106]
表17实施例8中化合物的产量如表格18所示:
[0107][0108][0109]
表18
[0110]
二、生物活性测试
[0111]
化合物1-12神经保护活性筛选
[0112]
1、采用cck8测定细胞活力
[0113]
细胞培养:c57bl/6孕鼠处死后取出胎脑,剥离血管膜和其他组织,得到大脑皮层,用0.5
‰
胰蛋白酶和0.1ku dnasei/ml消化,轻缓吹打,37℃消化约20min,每隔10min吹打一次,之后用10%fbs的dmem终止消化。将得到细胞离心(1000rpm,3~5min),重悬细胞,计数,用含10%fbs+1%链青霉素双抗的dmem培养基将细胞按适当的密度铺在96孔板中。所用细胞培养皿及细胞培养板均用多聚-l-赖氨酸预先包被过。37℃的培养箱培养4h后全量换液,换成神经元培养基,继续在5%co2的37℃的培养箱。之后每3天半量换液一次,第8天神经元即为成熟神经元,即可进行后续处理。
[0114]
配置cck8工作液:按照1:10的比例将cck8溶液与培养基混合,备用。现配现用。
[0115]
cck8孵育:从37℃细胞培养箱中取出96孔细胞培养板,吸弃原有培养基,将配好的cck8工作液按100μl/孔的量加入96孔板中。重新将96孔板放回细胞培养箱中孵育1~4h(孵育时间由od
450nm
约为1.0时决定)。
[0116]
od450 nm测定:用多功能酶标仪测定每孔的od
450nm
。将吸测定波长设置成450nm,测定前震荡10s以便混匀,测定吸光度。检测化合物对原代神经元细胞活力的影响。
[0117]
2、采用ogd/r观察化合物神经保护活性
[0118]
将上述处理好的神经元细胞进行氧糖剥夺-复氧复糖损伤,然后观察化合物的神经保护活性。具体操作如下:
[0119]
ogd/r:首先用95%n2-5%co2的混合气体饱和无糖dmem 15~30min,并37℃预热。弃去细胞原有培养基,用预先准备好的预热的不含氧的无糖培养基洗3遍,然后加入适量的无氧无糖dmem,放入37℃的三气培养箱中培养(94%n2,5%co2,1%o2)培养。2h后,换成细胞相应的正常培养基继续培养22h,进行后续cck8实验。
[0120]
测定450nm波长处的吸光度值,计算细胞存活率。细胞存活率=[(处理孔吸光度-空白孔吸光度)/(对照孔吸光度-空白孔吸光度)]
×
100%。
[0121]
3、神经保护
[0122]
化合物1-12用dmso溶解,并最终稀释到固定浓度10μm。实验采用神经元细胞进行氧糖剥夺-复氧复糖损伤(ogd/r)模型,评价化合物1-12对神经细胞的活度影响情况。
[0123]
ctrl组为未进行ogd/r操作,故细胞活度为100%。
[0124]
dmso为溶剂。
[0125]
阳性对照药物为:依达拉奉。
[0126]
化合物1~12、ctrl组、dmso、阳性对照物的神经保护细胞活度(即为细胞存活率)如表格19所示:
[0127][0128]
表19
[0129]
通过活性测试发现,化合物1~12均具有显著的神经保护活性,个别化合物与阳性对照药相当或更优。实施例1的化合物1~12的神经保护活性如图50所示。
[0130]
综上所述,本发明的化合物1~12均具有显著的神经保护活性,个别化合物与阳性对照药相当或更优。所以,本发明有效克服了现有技术中的种种缺点而具高度产业利用价值。
[0131]
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1