一种判断古DNA样本种族归属的方法
一种判断古dna样本种族归属的方法
技术领域
1.本发明属于生物技术中的生物工程技术领域,具体涉及一种基于多重 snps(多个单核苷酸多态性)表征古代dna样本继而判断样本的种族归属的方法。
背景技术:
2.满-通古斯语族分布在中国分布在黑龙江、内蒙古东北部和新疆;国外分布在俄罗斯东部、蒙古国。中国境内有5种语言:满语、锡伯语、赫哲语、鄂温克语、鄂伦春语以及历史上女真族使用的女真语。这些语言分布于黑龙江省的黑河市、逊克县、呼玛县、塔河县、富裕县、讷河县、嘉荫县、同江县、饶河县,内蒙古自治区的鄂温克族自治旗、鄂伦春自治旗、陈巴尔虎旗、莫力达瓦达斡尔族自治旗、阿荣旗、额尔古纳左旗、扎兰屯市,新疆维吾尔自治区的察布查尔锡伯自治县、霍城县、巩留县、塔城县、伊宁市、乌鲁木齐市等地。苏联境内有8 种语言:埃文基语、埃文语、涅基达尔语、那乃语、乌利奇语、奥罗克语、乌德盖语、奥罗奇语。这些语言分布于东西伯利亚和远东的埃文基自治专区、雅库特自治共和国、布里亚特自治共和国、哈巴罗夫斯克边疆区、滨海边疆区、萨哈林州、堪察加州、马加丹州等地。蒙古国境内只有鄂温克语,分布于巴尔虎地区。使用满-通古斯语言的民族总人口约445万。其中,中国约439万(1982),苏联约5.64万(1979),蒙古国1000多。
3.语言的产生是个漫长的过程。如果不同地域的人说的是同一类或同一种语言,则这些人群之间一定拥有漫长的共同进化的历史(除去殖民者或统治者通过行政手段促使人们使用指定的语言这一因素)。另外,如果不同地域的人种其语言基因的多态性模式如果很接近,也会存在很大的可能,即他们在远古的进化道路上有很深的渊源。
4.赫哲族是中国东北地区一个历史悠久的少数民族,民族语言为赫哲语,属阿尔泰语系满-通古斯语族满语支(也有观点认为应归入那乃次语支),没有本民族的文字,使用西里尔字母来记录语言,因长期与汉族交错杂居,通用汉语。由于居住地域广阔,赫哲人的自称较多,如“那贝”、“那乃”、“那尼傲”,“赫哲”作为族称最早出现于康熙二年(1663)三月,1934年凌纯声《松花江下游的赫哲族》一书出版后,“赫哲”作为族称开始广泛传播。赫哲族主要分布于黑龙江、松花江、乌苏里江交汇构成的三江平原和完达山余脉,集中居住于三乡两村,即同江市街津口赫哲族乡、八岔赫哲族乡、双鸭山市饶河县四排赫哲族乡和佳木斯市敖其镇敖其赫哲族村、抚远县抓吉镇抓吉赫哲族村。根据《中国统计年鉴-2021》,中国境内赫哲族的人口数为5373人。
5.鄂伦春这一族称,在1640年4月28日是以“俄尔吞”出现的。1683年(康熙二十二年)以后,文献中多次出现“俄罗春”、“鄂罗春”、“鄂伦春”等不同写法。从1690年(康熙二十九年)十月始,“鄂伦春”才作为统一的族称固定下来。“鄂伦春”是民族自称,即“使用驯鹿的人们”。另外,“鄂伦”的发音与驯鹿的发音(oron)相同,(cho)是表示人的附加成分,两者合起来为(oroncho),即“鄂伦春”,汉语就是“打鹿人”之意。十七世纪中叶以前,鄂伦春人分布于贝加尔湖以东、黑龙江以北,以精奇里江为中心的广大地区。鄂伦春族是世居我国东北部地区的人口最少的民族之一。根据《中国统计年鉴-2021》,鄂伦春族人口为9168人。鄂伦春语
属阿尔泰语系满—通古斯语族通古斯语支,没有文字,现在主要使用汉语汉文。
6.鄂温克族(旧称通古斯或索伦)是东北亚地区的一个民族,主要居住于俄罗斯西伯利亚以及中国内蒙古和黑龙江两省区,蒙古国也有少量分布。在俄国被称为埃文基人。鄂温克是鄂温克族的民族自称,其意思是“住在大山林中的人们”。鄂温克民族的语言文化具有独特性,属阿尔泰语系之通古斯语族北语支,在日常生活中,鄂温克人多数使用本民族语言,没有本民族的文字。鄂温克牧民大多使用蒙古文,农民则广泛使用汉文。鄂温克人是从游牧发展到定居的,从事畜牧业生产方式的人群。他们的传统文化具有极大的丰富性,最为突出的是服饰文化和饮食文化。根据《中国统计年鉴-2021》,中国境内鄂温克族的人口数为34617 人。
7.在人类进化研究中,本专利首次发现赫哲族和鄂伦春族与欧洲的尼安德特人(homo neanderthalensis简称尼人)在语言基因多态性模式上很接近甚至几乎完全一致,暗示该民族的现代和古代dna样本未来将具备重大的潜在研究价值。原因之一是,有证据表明,欧洲的尼人在许多特征上与北京人有雷同之处。尼人很可能来自中国的北方,尤其是黑龙江流域及附近的通古斯语族人种(包括赫哲族和鄂伦春族),尽管目前欧洲人倾向于认为欧洲的尼人通过在欧亚大陆上的长期迁徙并于东亚当地古人的杂交,成就了东亚的人类之进化。目前国际上公认尼人来自于非洲。由于欧洲地域相对狭小,应对冰河期寒冷气候的纵深空间很有限,导致尼人不得不向东亚迁徙甚至回迁到非洲。滞留于欧洲的尼人在冰河期间不断灭绝,而迁徙东亚或南下非洲的部分尼人及其后续进化的若干人种反而有机会存续下来。如果尼人来自东北亚,则人类起源于非洲的说法将几乎土崩瓦解,整个人类进化史将被改写。
8.人类的进化史可能还需要从更古老的地质年代找到答案,即:在更古老的地质年代,全球的主要大陆都是相连的,某一类远古猿类其实是最早的共同祖先;之后大陆分离成为几个大洲,各个大洲的古猿分别进化但是有相同的基因组基础,保证了各个地域演化的古人类具有相同的科属。地质冰川时期的周期性循环使得各个大洲的宏观气候特征类似,保证了各个陆地古猿之间的适量互动和杂交,使得各地古猿的进化方向大抵一致,直到不止一个地方同时出现了直立人(但是已经存在地域性差异)。这些直立人之间一直并不存在大的生殖隔离,且不同进化阶段的直立人同时存在。后面起关键作用的是局部气候决定了哪些地方的直立人或智人率先出走,带动了后续的人种之快速融合和多域进化及局部消亡。
9.基于文献研判得到的总的结果是:(1)国际上目前由西方主导的研究表明,人类起源于非洲;古猿大约200万年前演化成直立人;古人类(直立人)走出非洲,先是到达欧洲和西亚,之后进入南亚和东亚,继而进入东南亚和澳洲,然后在大约12000年前进入北美,继而延伸到南美。(2)中国主导的研究(大部分结果没有以英文发表,不为西方所知)有很强的证据表明,猿猴最早起源于中国西南,古猿进化到直立人,继而产生智人等各个阶段在中国都有大量的化石证据;中国西南地区214万年前就存在巫山人;更早期的化石将来应该也会发现。(3)文献资料表明,存在下列可能:古地质时期由于欧亚大陆与非洲连在一起,那时候进化的古猿有可能从中国西南迁徙或自然扩散到非洲等地,从而保证中国西南和非洲东部进化成直立人的那部分古猿具备相同的祖先,即具备相同的起始基因组,这保证了之后在中国西南和非洲东部的直立人及其进化的后裔在基因组层面属于相同的科属;(4)东非大裂
谷的形成时间与中国西南喜马拉雅山脉的第二次隆起时间段比较吻合,都是大约在500-200万年前。两个地点的地貌巨变给当地的古猿带来了类似的挑战,即局部地域的森林消退,古猿不得不适应稀树草原(东非)或河流峡谷(中国西南),两个地点都需要古猿尽快具备直立行走的能力才能生存;(5)只要直立人不具备长期保存火种或主动制备火种的能力并同时具备制备兽衣抵御严寒的能力,将无法在一次次冰河期间得以存活,很容易在森林、山洞或草原中失温而亡。从这个角度来讲,中国西南地域地貌复杂,更容易帮助古猿和各个进化阶段的古人类躲过冰期严寒,比非洲更容易满足存续条件;(6)世界各个地方的直立人或其后续进化人种在每次冰河期间未必都会全部死亡,更有可能是各种古人都会小部分存活下来(当然不排除有的人种全部消亡);由于不存在大的生殖隔离,几乎所有这些古人种都可以杂交融合产生新的人种,这保证了人类在进化过程中基因组序列的连续性和高度相似性。
10.尼安德特人(homo neanderthalensis),简称尼人,常作为人类进化史中间阶段的代表性居群的通称,属于智人的一种。因其化石发现于德国尼安德特山谷而得名。尼安德特人分布很广,西起欧洲的西班牙和法国,东到中亚的乌兹别克斯坦,南到巴勒斯坦,北到北纬53
°
线。年代最早的距今达20万年左右,最晚的距今约4万年。尼安德特人是现代欧洲人祖先的近亲,从12万年前开始,他们统治着整个欧洲、亚洲西部以及非洲北部,但在两万四千年前,这些古人类却消失了。2010年,尼安德特人基因组草图发布,也基于尼安德特人基因组草图,研究结果发现,除非洲人之外的欧亚大陆现代人均有1%~4%的尼安德特人基因成分贡献。2017年,3月3日美国《科学》杂志3日发表题为《中国许昌出土晚更新世古人类头骨研究》论文称,人类演化研究取得突破性进展:10多万年前生活在河南省许昌市灵井遗址的“许昌人”,可能是中国境内古老人类和欧洲尼安德特人的后代。
11.语言能力的进化过程如何匹配人类的总的进化过程是一个重要的课题。可以肯定的是,语言能力应该是在直立人期间就得到了重大的发展,智人阶段应该进一步将语言能力加以提升和分化,而晚期智人的语言能力应该与现代人无异了。目前的研究发现(1)除了非洲外,世界各地现代人的语言基因多态性模式似乎极为接近;(2)有个别的丹尼索瓦人和尼安德特人居然拥有与现代人几乎非常接近的语言基因多态性模式,这暗示丹尼索瓦人和尼安德特人应该可以像现代人一样说话和发音;(3)现代非洲人的语言基因多态性模式与其他国家地域差别很明显,有一些国家如西班牙和肯尼亚的语言基因模式介于非洲人与其他国家现代人之间。
12.在人类进化的研究过程中,古dna一直是一个利器。通过对远古dna或化石dna的提取和精细测序,结合分子遗传学的定量方法,可以比较精确地推断对应人种与其他已知基因组序列的人种之间之进化距离。测序分为全基因组测序和部分区段测序。很多远古dna由于含量稀少且支离破碎,很难从这样的样本中得到全基因组的序列;当然部分区段测序结果也可以很好地预测进化距离或亲缘关系。但是,当同时比较大量复杂样本之间的亲缘关系时,总会受到到算法的限制而难以对有些样本进行亲缘关系的精确定位。本发明通过一组固定的数量较为庞大的语言基因多态性参数和一组已知进化距离或亲缘关系的标准dna序列样本,可以较好地对赫哲族或鄂伦春族的古dna样本进行种族归属的定位或辅助定位。
技术实现要素:
13.本发明的目的在于进一步完善现有技术,提供一种基于语言基因多态性 snps标记的古dna种族确定方法;通过提取古代样本中的古dna,对古dna直接建库并实施全基因组高通量测序,从所测序列中收集13个语言基因的147个单核苷酸多态性位点信息,对这些位点信息数字化表征并与一组对照序列做主成分分析(pca),继而确定古dna样本是否属于赫哲族或鄂伦春族。
14.本发明的技术路线
15.(1)古dna的提取;一般采用如下文献中的方法即可:dna analysis of an early modern human from tianyuan cave,china.proc natl acad sci usa.2013feb 5; 110(6):2223-7.也可以根据样本的性质和来源采取适当的其他已知方法;
16.(2)全基因组高通量测序:采用二代高通量测序等适合古dna的全基因组测序方法,得到fastq等格式的基因组序列文件;
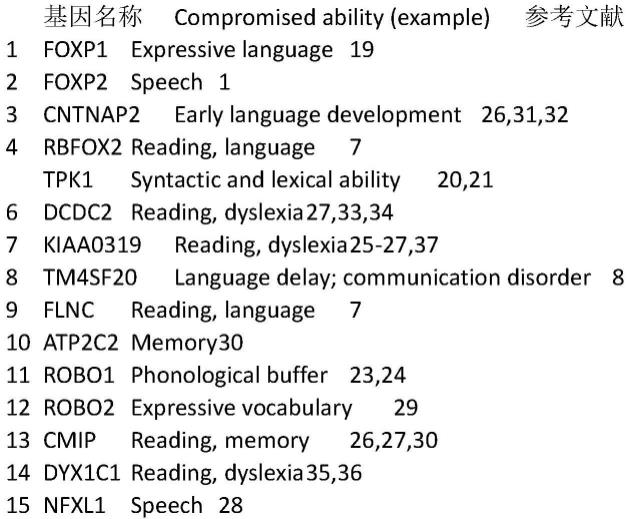
17.(3)语言基因多态性snps:涉及的13个语言基因为foxp1,foxp2,cntnap2, rbfox2,tpk1,dcdc2,kiaa0319,tm4sf20,flnc,atp2c2,robo1,robo2, cmip,dyx1c1,nfxl1;每个基因采用4-12个snp位点,总共147个snp位点。所采用的snp位点以尽可能具有特定地域特异性为准。采用的位点越多,则样本之间的细微差别就更容易被识别,古dna的种族定位做的就越精准;
18.(4)对照序列:目前国际上公认人类起源于非洲;古猿大约200万年前演化成直立人;古人类(直立人)走出非洲,先是到达欧洲和西亚,之后进入南亚和东亚,继而进入东南亚和澳洲,然后在大约12000年前进入北美,继而延伸到南美。这期间发生过2-3次走出非洲的时间,对应着不同阶段的古人类(直立人、智人、现代人等)。也发生过backtoafrica的事件,但是具体多少次尚不清楚。尤其是几个主要的大陆之间发生过多少次主要的人类迁徙也需要今后多年的研究。但是要对未知古dna样本进行种族定位,一般需要包括如下几个洲或地域的对照序列:非洲、美洲、欧洲、东亚、南亚、东南亚、大洋洲等的古dna和现代dna。世界上很多地区的古dna研究刚刚开始,目前缺乏足够的高质量的古dna,但是非洲、欧洲、亚洲的古dna是必需的对照序列。
19.(5)pca分析:采用r语言中的factominer程序包及其辅助程序包实施pca 分析。通过pca输出的精细图中的位置以及对照序列的相对位置来判定或辅助判定未知古dna是否属于赫哲族或鄂伦春族。
20.本发明的优点和效果
21.1、特定的snp组合:
22.与语言功能直接相关的基因称之为语言基因。基因一般由agct四种碱基组成.一段基因序列的特定位置在不同个体身上表现出来的单个碱基的差异被称为单核苷酸多态性(single nucleotide polymorphism,snp)。比如某基因的特定位置在不同的人群中有的是a,有的是t,那么该位置就是该基因的一个a/t多态性位点. 语言基因的多态性是不同个人语言能力有明显差异的遗传基础。在不同种族、不同地域、不同个体上有其不同的分布特点。因为语言基因本身的个数就至少有十几个,每个基因的相关snp数目也可以多达几个到几十个,而且语言基因需要与其他至少几十个基因相互作用(还需要与环境进一步相互作用)才能最终决定个体语言能力的具体性状,所以可以想象不同的个体之间语言能力的生
物学基础之差异应该是非常大的。这也是不同种族不同地域个体之间语言功能相关表型多样化的分子基础。
23.表格1本专利涉及的语言基因名单
[0024][0025]
说明:这15个人类基因都有对应的实验证据说明与语言功能有关。
[0026]
参考文献:
[0027]
[1]laicsetal.afork-headdomaingeneismutatedinaseverespeechandlanguagedisorder.nature,2001,413(6855):519-523.
[0028]
[7]gialluisiaetal.genome-widescreeningfordnavariantsassociatedwithreadingandlanguagetraits.genesbrainandbehavior,2014,13(7):686-701.
[0029]
[8]wiszniewskiwetal.tm4sf20ancestraldeletionandsusceptibilitytoapediatricdisorderofearlylanguagedelayandcerebralwhitematterhyperintensities.americanjournalofhumangenetics,2013,93(2):197-210.
[0030]
[19]baconc&garappold.thedistinctandoverlappingphenotypicspectraoffoxp1andfoxp2incognitivedisorders.humangenetics,2012,131(11):1687-9168.
[0031]
[20]villanuevapetal.genome-wideanalysisofgeneticsusceptibilitytolanguageimpairmentinanisolatedchileanpopulation.europeanjournalofhumangenetics,2011,19(6):687-695.
[0032]
[21]fattalietal.thecrucialroleofthiamineinthedevelopmentofsyntaxandlexicalretrieval:astudyofinfantilethiaminedeficiency.brain,2011,134(6):1720-1739.
[0033]
[23]hannula-jouppi,k.etal.theaxonguidancereceptorgenerobo1isacandidategenefordevelopmentaldyslexia.plosgenet.1,e50;(2005).
[0034]
[24]bates,t.c.etal.geneticvarianceinacomponentofthelanguageacquisitiondevice:robo1polymorphismsassociatedwithphonologicalbufferdeficits.behav. genet.41,50
–
57;(2011).
[0035]
[25]paracchini,s.etal.thechromosome6p22haplotypeassociatedwithdyslexiareducestheexpressionofkiaa0319,anovelgeneinvolvedinneuronalmigration.hum.mol.genet.15,1659
–
1666;(2006).
[0036]
[26]newbury,d.f.etal.investigationofdyslexiaandsliriskvariantsinreading-andlanguage-impairedsubjects.behav.genet.41,90
–
104;(2011).
[0037]
[27]scerri,t.s.etal.dcdc2,kiaa0319andcmipareassociatedwithreading-relatedtraits.biol.psychiatry70,237
–
245;(2011).
[0038]
[28]villanueva,p.etal.exomesequencinginanadmixedisolatedpopulationindicatesnfxl1variantsconferariskforspecificlanguageimpairment.plosgenet.11,e1004925;(2015).
[0039]
[29]stpourcain,b.etal.commonvariationnearrobo2isassociatedwithexpressivevocabularyininfancy.nat.commun.5,4831;(2014).
[0040]
[30]newbury,d.f.etal.cmipandatp2c2modulatephonologicalshort-termmemoryinlanguageimpairment.am.j.hum.genet.85,264
–
272;(2009).
[0041]
[31]vernes,s.c.etal.afunctionalgeneticlinkbetweendistinctdevelopmentallanguagedisorders.n.engl.j.med.359,2337
–
2345;(2008).
[0042]
[32]whitehouse,a.j.,bishop,d.v.,ang,q.w.,pennell,c.e.&fisher,s.e.cntnap2variantsaffectearlylanguagedevelopmentinthegeneralpopulation.genesbrainbehav.10,451
–
456;(2011).
[0043]
[33]deffenbacher,k.e.etal.refinementofthe6p21.3quantitativetraitlocusinfluencingdyslexia:linkageandassociationanalyses.hum.genet.115,128
–
138;(2004).
[0044]
[34]schumacher,j.etal.stronggeneticevidenceofdcdc2asasusceptibilitygenefordyslexia.am.j.hum.genet.78,52
–
62;(2006).
[0045]
[35]taipale,m.etal.acandidategenefordevelopmentaldyslexiaencodesanucleartetratricopeptiderepeatdomainproteindynamicallyregulatedinbrain.proc.natl.acad.sci.usa100,11553
–
11558;(2003).
[0046]
[36]paracchini,s.etal.analysisofdyslexiacandidategenesintherainecohortrepresentingthegeneralaustralianpopulation.genesbrainbehav.10,158
–
165;(2011).
[0047]
[37]francks,c.etal.a77-kilobaseregionofchromosome6p22.2isassociatedwithdyslexiainfamiliesfromtheunitedkingdomandfromtheunitedstates.am.j.hum.genet.75,1046
–
1058;(2004).
[0048]
表格2语言基因的147个snp位点
[0049]
genesnpnamesnpabbreviation
[0050]
atp2c2rs78371901,rs4782948,rs2435172,rs247885,rs247818,rs74038217,rs62640935,rs62640932,rs62640931,rs62050917,rs16973859,rs13334642,rs4782970atp-1,atp-10,atp-11,atp-12,atp-13,atp-2,atp-3,atp-4,atp-5,atp-6,atp-7,atp-8,atp-9
[0051]
cmip rs201316817,rs34119643,rs16955675,rs2288011,rs1187121850, rs183876152,rs183075361,rs114894868,rs79979027,rs74031247,rs60152409, rs57603843,rs35429777 cmi-1,cmi-10,cmi-11,cmi-12,cmi-13,cmi-2, cmi-3,cmi-4,cmi-5,cmi-6,cmi-7,cmi-8,cmi-9
[0052]
cntnap2 rs1637842,rs3194,rs535454043,rs2373284,rs61732853, rs1637841,rs1479837,rs1468370,rs1062072,rs1062071,rs987456,rs700309, rs700308 cntn-1,cntn-10,cntn-11,cntn-12,cntn-13,cntn-2,cntn-3, cntn-4,cntn-5,cntn-6,cntn-7,cntn-8,cntn-9
[0053]
dcdc2 rs35029429,rs33914824,rs33943110,rs190254728,rs2274305, rs34584835,rs33943110,rs33914824,rs9467075,rs9460973,rs3846827,rs3789219
[0054]
dcd-1,dcd-10,dcd-11,dcd-12,dcd-2,dcd-3,dcd-4,dcd-5,dcd-6, dcd-7,dcd-8,dcd-9
[0055]
flnc rs2291569,rs2249128,rs117864464,rs35281128,rs371111092, rs2291568,rs2291566,rs2291565,rs2291563,rs2291562,rs2291561,rs2291560, rs2291558 fln-1,fln-10,fln-11,fln-12,fln-13,fln-2,fln-3,fln-4,fln-5, fln-6,fln-7,fln-8,fln-9
[0056]
foxp1 rs76145927,rs17008224,rs147756430,rs75214049,rs17008544, rs17008063,rs11914627,rs7639736,rs1499893,rs1053797,rs144080925
[0057]
foxp1-1,foxp1-10,foxp1-11,foxp1-2,foxp1-3,foxp1-4,foxp1-5,foxp1-6, foxp1-7,foxp1-8,foxp1-9
[0058]
foxp2 rs10227893,rs144807019,rs182138317,rs61758964,rs10244649, rs12705977,rs61732741,rs61758964,rs62640396,rs73210755,rs1058335, rs61753357,rs7638391 foxp2-1,foxp2-10,foxp2-11,foxp2-12,foxp2-2, foxp2-3,foxp2-4,foxp2-5,foxp2-6,foxp2-7,foxp2-8,foxp2-9,fxp1
[0059]
kiaa0319 rs138160539,rs75720688,rs150584710,rs115399701,rs7770041, rs117692893,rs114195393,rs699461,rs699462,rs699463,rs730860,rs10946705, rs75674723 kia-1,kia-10,kia-11,kia-12,kia-13,kia-2,kia-3,kia-4,kia-5, kia-6,kia-7,kia-8,kia-9
[0060]
nfxl1 rs1964425,rs920462,rs147017712,rs13152765,rs34323060, rs1822030,rs1822029,rs1812964,rs1545200,rs1440228,rs1371730,rs1036681, rs978094 nfx-10,nfx-11,nfx-12,nfx-13,nfx-2,nfx-3,nfx-4,nfx-5, nfx-6,nfx-7,nfx-8,nfx-9
[0061]
robo1 rs34841026,rs77350918,rs6795556,rs35456279
[0062]
robo-10,robo-14,robo-15,robo-16
[0063]
robo2
[0064]
rs11127602,rs78817248,rs144468527,rs17525412,rs10865561,rs5788280,rs392 3745,rs3923744,rs1163750,rs1163749,rs1163748,rs1031377
[0065]
robo-1,robo-11,robo-12,robo-13,robo-2,robo-3,robo-4,robo-5,robo-6, robo-7,robo-8,robo-9
[0066]
tm4sf20
[0067]
rs6724955,rs137891000,rs44675173,rs4675172,rs4673192,rs4438464,rs442801 0,rs4408717,rs13415654,rs80305648
[0068]
tm1,tm10,tm2,tm3,tm4,tm5,tm6,tm7,tm8,tm9
[0069]
tpk1
[0070]
rs113536847,rs77358162,rs79464600,rs77358162,rs28380423,rs17170295,rs12 333969,rs6953807,rs67644764
[0071]
tpk-1,tpk10,tpk-2,tpk-3,tpk-4,tpk-5,tpk-6,tpk-7,tpk-8
[0072]
2、特定的对照序列:
[0073]
本发明提供的对照序列包括非洲、美洲、欧洲、东亚、南亚、东南亚、大洋洲的现代人基因组序列以及东亚、欧洲及非洲的一组古dna基因组序列,见表格3,含有3个待测样本和73个对照样本.
[0074]
表格3特定的对照序列
[0075]
[0076]
[0077][0078]
3、特定的pca计算方法:
[0079]
本发明采用r语言平台提供的一种pca分析技术,采用factominer程序包以便处理具有不同单位不同大小范围变量的归一化。所有的snp位点信息采用如下方法进行数字化处理:1000*a frequency-1000*t frequency-1000*g frequency-1000*c frequency;比如如果一个snp的位点信息是a=0,t=0.166, g=0.834,c=0,则量化为166834000;而a=0.166,t=0,g=0,c=0.834,则量化为 166000000834;如果某个碱基的频率是1,则量化为999,比如,a=0,t=1,g=0, c=0量化为999000000;a=1,t=0,g=0,c=0量化为999000000000;在语言基因的 snp信息收集过程中,会发现同一个样本的基因组序列中某一个snp位点既有含有a的序列也有含有c的序列,即a=1,t=0,g=0,c=1,则量化为999000000999;如果某个语言基因snp位点测定为a=0,t=1,g=0,c=1,则量化为999000999;a=1, t=0,g=1,c=1,则量化为999000999999;
具体实施方式
[0080]
1、古dna的提取:使用商业化古dna提取试剂qiaquickr试剂盒)对古 dna进行提取,具体步骤如下:
[0081]
(1)称取0.1g样本,加入1ml10%sds、4ml0.5m edta、100μl10mg/ml蛋白酶k,50℃振荡孵育24h;
[0082]
(2)将裂解液8000r/min离心8min;
[0083]
(3)每次取450μl上清液加入到centriconym-10中,6500r/min离心,重复多次,最终把上清液浓缩到100μl;
[0084]
(4)加入5倍体积的qiaquickbufferpb,再加2μl phi混合30s;
[0085]
(5)加入到qiaquickspincolumn中,13000r/min离心1min;
[0086]
(6)倒掉滤液,13000r/min离心1min;
[0087]
(7)加入500ul bufferpe,13000r/min离心1min,总共洗涤2次;
[0088]
(8)再13000r/min离心3min,使过滤器干燥;
[0089]
(9)加入100μl的buffereb,53℃孵育8min,13000r/min离心2min,收集滤液,-20℃储存。
[0090]
(10)采用thermonanodrop2000分光光度计对试剂盒法提取出的古dna 溶液进行浓度和纯度测定。
[0091]
依据不同的样本来源和样本量,也可以参照如下文献方法进行古dna的提取:一种皮张中古dna的提取方法及试剂盒cn201810465329.3;一种木质文物的古dna捕获方法cn202210149079.9;一种古dna文库构建的方法 cn202110894520.1;一种高效的古代dna提取方法cn202110652184.x;一种针对dna含量极低的古代人类遗骸性别鉴定方法
cn201910185048.7;一种担载sio2磁珠快速提取古dna的方法cn201310675886.5;鉴定和分析古dna样本的方法cn201710667605.x;或dnaanalysisofanearlymodernhumanfromtianyuancave,china.procnatlacadsciusa.2013feb5;110(6):2223-7.
[0092]
2、高通量测序:古dna不需要进行碎片化处理,直接进行文库构建,末端修饰、接头反应,扩增,微流体池反应,信号数据采集得到rawdata;利用相关平台软件处理rawdata并得到全基因组序列的fastq等格式的基因组序列文件;
[0093]
3、snp位点信息获取及信息数字化:
[0094]
针对13个语言基因的总共147个位点,在dbsnpdatabase:https://www.ncbi.nlm.nih.gov/snp/中下载每个snp的序列信息(dbsnp展示的基本上都是现代人类的snp信息;所以本专利本质上是利用现代人类的语言基因多态性信息来判断不同人类基因组样本之间的相对进化距离,继而实现对特定样本之进化分类归属的判断或辅助判断),然后使用位点前后各18-25个碱基的序列在fastq序列文件中寻找snp的具体碱基信息。fastq基因组序列文件一般是越大越好,但是限于电脑的内存容量,可以使用2g-200g大小的fastq文件进行snp信息检索。比如,如果200g这个大小超过计算机内存,可以把文件分成2个小一点的文件分别检索。古dna的基因组序列经常因为样本历史久远降解严重而只有几个g的大小,但是很多对照序列的古基因组信息得到了高质量的测序,序列总大小可以达到200g以上。全部147个snp位点的选择主要依托1000genomes/ensemblorgnomadgenomesr3.0这些数据库中的信息,最核心的选择依据是所选的snp尽量在不同的种族中出现频率有明显差别,利用这样的snp有利于后续对不同样本进行区分。所有的snp位点信息采用如下方法进行数字化处理:1000*afrequency-1000*tfrequency-1000*gfrequency-1000*cfrequency;比如如果一个snp的位点信息是a=0,t=0.166,g=0.834,c=0,则量化为166834000;而a=0.166,t=0,g=0,c=0.834,则量化为166000000834;如果某个碱基的频率是1,则量化为999,比如,a=0,t=1,g=0,c=0量化为999000000;a=1,t=0,g=0,c=0量化为999000000000;在语言基因的snp信息收集过程中,会发现同一个样本的基因组序列中某一个snp位点既有含有a的序列也有含有c的序列,即a=1,t=0,g=0,c=1,则量化为999000000999;如果某个语言基因snp位点测定为a=0,t=1,g=0,c=1,则量化为999000999;a=1,t=0,g=1,c=1,则量化为999000999999;
[0095]
4、pca计算:主成分分析principalcomponentmethods(pca)用于从多变量数据表中提取重要信息,并将此信息表示为一组称为主成分的新变量。这些新变量对应于原始变量的线性组合。主成分的数量小于或等于原始变量的数量。pca的目标是识别数据变化最大的方向(或主成分),将多变量数据的维度降低到两个或三个主要成分,这些成分可以图形化可视化,同时信息损失最小。pca属于机器学习降维方法质之一,但是仅仅对线性数据有用,非线性数据建议使用tsne。本专利中涉及的snp数据属于线性数据。总的来说主成分分析的主要目的是识别数据集中的隐藏模式;通过消除数据中的噪声和冗余来降低数据的维度;识别相关变量。在主成分分析中变量通常被缩放(即标准化)。当变量以不同的尺度(例如:千克,千米,厘米
…
)被测量时,尤其需要这样做;否则获得的pca输出将受到严重影响,其目标是使变量具有可比性。通常,变量被缩放为具有标准偏差1和平均值为零。数据标准化是在pca和聚类分析之前广泛用于基因表达数据分析的方法。默认情况下在r语言的程序包factominer中,pca之前会自动标准化数据;在r包中,factominer包从探索性分析的角
度(对数据集进行描述、绘制并可视化)对传统的多元统计方法进行了扩展,包括如下方法1)降维方法:主成分分析(pca)、因子分析(fa,包括多重因子分析mfa、层次多重因子分析hmfa以及混合数据因子分析famd)、对应分析(ca,包括多重对应分析mca)(2)聚类分析方法:层次聚类、k-均值聚类和基于模型的聚类。 factominer很好地整合了多元分析的结果,还具有如下特点:可以考虑不同类型的变量(定量或分类)、不同类型的数据结构(变量划分、变量层次结构、个体划分)以及补充信息(补充个体和变量)。factoextra包是factominer的一个补充。它使用factominer的计算结果,在另一个r包ggplot2的基础上给出多元分析结果更美观的可视化。具体的计算及可视化过程如下:
[0096]
》library(factominer)
[0097]
》library(factoextra)
[0098]
》library(ggplot2)
[0099]
》country《-read.delim('c:/rbook/20220516fastqsnpdata.txt',row.names=1, sep='\t')
[0100]
》country《-t(country)
[0101]
》country.pca《-pca(country,ncp=2,scale.unit=true,graph=false)
[0102]
》plot(country.pca)
[0103]
》pca_sample《-data.frame(country.pca$ind$coord[,1:2])
[0104]
》head(pca_sample)
[0105]
》pca_eig1《-round(country.pca$eig[1,2],2)
[0106]
》pca_eig2《-round(country.pca$eig[2,2],2)
[0107]
》pca_eig1
[0108]
》pca_eig2
[0109]
》group《-read.delim('c:/rbook/group3.txt',row.names=1,sep='\t', check.names=false)
[0110]
》group《-group[rownames(pca_sample),]
[0111]
》pca_sample《-cbind(pca_sample,group)
[0112]
》pca_sample$samples《-rownames(pca_sample)
[0113]
》head(pca_sample)
[0114]
》library(ggrepel)
[0115]
》ggplot(data=pca_sample,aes(x=dim.1,y=dim.2))+geom_point(aes(color =group),size=3)+scale_color_manual(values=c('purple','red', 'green','blue','brown','pink','yellow','orange','grey'))+theme(panel.grid= element_blank(),panel.background=element_rect(color='black',fill= 'transparent'),legend.key=element_rect(fill='transparent'))+labs(x= paste('pca1:',pca_eig1,'%'),y=paste('pca2:',pca_eig2,'%'),color=”)+ geom_text_repel(aes(label=samples),size=3,show.legend=false,box.padding= unit(0.25,'lines'))
[0116]
*说明:样本分组的个数与颜色种类相对应;颜色种类可以在orange,grey, yellow,dark,black,pink,green,brown,blue等颜色库中选取。
[0117]
表1本专利涉及的语言基因名单
[0118]
表2语言基因的147个snp位点
[0119]
表3特定的对照序列
附图说明
[0120]
图1赫哲族(古dna及现代dna)与鄂伦春族(现代dna)与特定尼人出现在pca图的特定区域。古dna样本(dvi,赫哲族)在pca分析中显示在尼人(nd2 和nd3)附近;dna样本(c4,赫哲族)在pca分析中显示在尼人(nd1和nd5)附近;dna样本(c5,鄂伦春族)在pca分析中显示在尼人(nd2和nd3)附近;实施例案例1对照样本(见表格3)+一组样本(亦见表格3);判断样本的种族归属,如果在pca图的最左侧位置且位于nd1-nd2-nd3-nd5这几个尼人的位置附近,则可以辅助判断为赫哲族或鄂伦春族来源的dna样本。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1