氟伐他汀钠关键中间体的合成方法与流程
1.本发明属于化学合成技术领域,具体涉氟伐他汀钠关键中间体的合成方法。
背景技术:
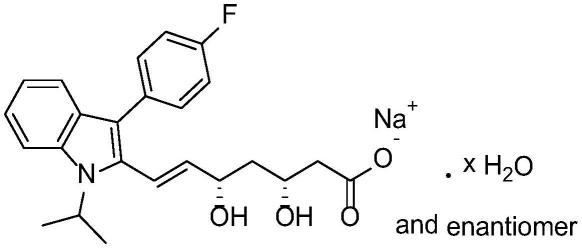
2.氟伐他汀钠(fluvastatin sodium)是由瑞士sandoz公司(现诺华制药公司)研制的首个全合成的甲基戊二酰辅酶a(hmc-coa)还原酶抑制剂类降血脂药物,于 1993年12月通过美国fda批准,次年于英美上市,1994年fda批准其胶囊上市, 2001年批准其缓释片剂上市,氟伐他汀具有结构相对简单、作用具有选择性和不良反应发生率低等优点,是一种优良的降血脂药。氟伐他汀能直接抑制肝脏的hmg-coa还原酶,且其体内的羟基代谢物仍有抑酶作用,用于治疗饮食调节无效的原发性高胆固醇血症和原发性混合型血脂异常。1997年,氟伐他汀钠被引入中国,商品名为来适可,现已进入中国国家医保名单,被喻为国产他汀类最具有潜力的产品之一。氟伐他汀钠的化学结构式如下:
[0003][0004]
式i,ii,iii化合物均可作为合成氟伐他汀钠的关键中间体,其结构如下:
[0005][0006]
美国专利us5354772a公开了氟伐他汀钠关键中间体,式i化合物的合成方法,合成路线如下:
[0007][0008]
该方法用[3'-(4"-氟苯基)-1'-异丙基-1h-吲哚-2"-基]-2-丙烯醛为原料,与乙酰乙酸甲酯在强碱作用下制备中间体i。该方法中采用了钠氢、正丁基锂,工艺危险性大,同样不利于大规模产业化应用。
[0009]
中国专利cn1217930c公开了氟伐他汀钠关键中间体,式i化合物的合成方法,合成路线如下:
[0010][0011]
该工艺采用侧链分段构建策略,首先吲哚化合物2-位进行vilsmeier-haack反应得到醛类中间体,再在正丁基锂等强碱作用下发生亲核取代等反应分段构建侧链。该工艺反应步骤长,总收率低,并且采用了钠氢、正丁基锂,工艺危险性大,缩合反应在零下78℃下反应,反应条件苛刻,不适合工业化生产。
[0012]
中国专利cn1217930c公开了氟伐他汀钠关键中间体,式i化合物的合成方法,合成路线如下:
[0013][0014]
该工艺以4-卤代乙酰乙酸酯为起始物料,先制备磷叶立德试剂,与吲哚醛化合物发生wittig反应构建二羰基侧链,随后在noyori催化剂下发生不对称氢化反应构建手性醇,再在强碱作用下发生亲核取代反应
[0015]
该工艺反应步骤长,总收率低,需引入大量磷试剂,环境污染较大,反应条件苛刻,需在零下78℃下反应,并且需用到贵金属钌,生产成本较大。
技术实现要素:
[0016]
本发明为解决现有技术中存在的不足,提供一种绿色高效、成本低廉,易于工业化应用的合成式i化合物的方法。
[0017]
根据本发明的目的,提供一种合成氟伐他汀钠中间体的方法,该采用有机锌试剂与(e)-3-[3'-(4"-氟苯基)-1'-异丙基-1h-吲哚-2"-基]-2-丙烯醛在双氮配体作用下合成得到如式i的氟伐他汀钠关键中间体,化学反应方程式如下:
[0018][0019]
所述有机锌试剂是通过α-溴代乙酸酯与活性锌反应得到,卤代物与活性锌的摩尔比为1∶1.5~1∶2.5,优选为1∶1.8~1∶2;其中活性锌是锌粉与四甲基氯硅烷(tmscl)活化反应得到的,活化过程可在制备锌试剂过程中同步进行,化学反应方程式如下:
[0020][0021]
所述合成方法中有机锌试剂与α-溴代乙酸酯的摩尔比为1∶3~1∶5,优选为 1∶4.5~1∶5。
[0022]
所述合成方法中双氮配体为n-甲基咪唑,二异丙基乙基胺,1,8-二氮杂双环 [5.4.0]十一碳-7-烯,2,2'-联吡啶,邻二氮菲,n,n-二甲基乙醇胺,四甲基乙二胺, n,n
’‑
二甲基乙二胺,优选四甲基乙二胺。其中双氮配体与(e)-3-[3'-(4"-氟苯基)-1'
‑ꢀ
异丙基-1h-吲哚-2"-基]-2-丙烯醛的摩尔比为1.5:1~2.5:1,优选为2:1。
[0023]
所述合成方法中碘盐催化剂为碘化钠,碘化钾,碘化镁,碘化铯,碘化亚铜,碘化锌,优选碘化亚铜。其中碘盐催化剂与(e)-3-[3'-(4"-氟苯基)-1'-异丙基
ꢀ‑
1h-吲哚-2"-基]-2-丙烯醛的摩尔比为0.05:1~0.5:1,优选为0.2:1。
[0024]
所述合成方法反应温度为0~80℃,优选反应温度50~60℃。
[0025]
所述合成方法中使用的反应溶剂为n,n-二甲基甲酰胺、二甲基亚砜、n, n-二甲基乙酰胺、1,3-二甲基-3,4,5,6-四氢-2-嘧啶酮、四氢呋喃,优选n, n-二甲基甲酰胺和二甲基亚砜。
[0026]
反应完毕,冷却到10-25℃,使用乙酸乙酯稀释,随后反应液依次使用20%柠檬酸水溶液,10%氯化钠水溶液,5%碳酸氢钠水溶液和自来水洗涤,静置分层后,分离有机相,40-50℃减压浓缩有机相,随后用10倍量的正庚烷和5倍量乙酸乙酯混合液重结晶得到产
物,收率为65-90%,纯度大于98.5%。
[0027]
现有技术中:文献及专利报道的合成式i化合物方法,使用了极低的反应温度-20℃和-78℃,且使用强碱,不但增加了产业化的风险,而且增加了后处理的难度,且会产生大量废水,不利于环保,此外,收率较低,增加了产业化的成本,不符合现今绿色化工的环保要求。
[0028]
本发明合成式i化合物具有显著的优点:使用有机锌试剂与(e)-3-[3'-(4"-氟苯基)-1'-异丙基-1h-吲哚-2"-基]-2-丙烯醛在双氮配体作用下合成化合物i,有机锌试剂使用锌粉与四甲基氯硅烷(tmscl)活化并与α-溴代乙酸酯通过简单的加热即可制得,可在制备过程中直接活化,不需分离可直接使用,反应过程温和,不需极低的反应温度,且后处理方便简单,常规的萃取水洗分层操作,避免了大量废水的产生,更加绿色环保;原料易得,产物收率高,最高可达90%以上,成本低廉,因此,更适合产业化。
具体实施方式
[0029]
下面结合具体实施例对本发明的实施方案进行详细描述,但是本领域技术人员应理解,实施例仅用于说明本发明,而不应视为限定本发明的范围。
[0030]
实施例中未注明具体条件者,按照常规条件、制造商或供应商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
[0031]
实施例1有机锌试剂brznch2co2me的合成
[0032]
氮气保护下,向1.0l反应瓶中投入锌粉57.5g(0.88mol),四氢呋喃220ml,四甲基氯硅烷4.8g(44mmol),升温至40-50℃,随后缓慢滴加α-溴代乙酸甲酯67.3g (0.44mol)的四氢呋喃溶液(500ml),继续搅拌反应1-3小时,反应完毕,静置(或离心)分离,澄清液收集后,有机锌试剂制备收率按100%计算,继续用于后续反应。
[0033]
实施例2有机锌试剂brznch2co2et的合成
[0034]
氮气保护下,向1.0l反应瓶中投入锌粉57.5g(0.88mol),四氢呋喃220ml,四甲基氯硅烷4.8g(44mmol),升温至40-50℃,随后缓慢滴加α-溴代乙酸乙酯73.5g (0.44mol)的四氢呋喃溶液(500ml),继续搅拌反应1-3小时,反应完毕,静置(或离心)分离,澄清液收集后,有机锌试剂制备收率按100%计算,继续用于后续反应。
[0035]
实施例3有机锌试剂brznch2co
2-i-pr的合成
[0036]
氮气保护下,向1.0l反应瓶中投入锌粉57.5g(0.88mol),四氢呋喃220ml,四甲基氯硅烷4.8g(44mmol),升温至40-50℃,随后缓慢滴加α-溴代乙酸异丙酯 79.6g(0.44mol)的四氢呋喃溶液(500ml),继续搅拌反应1-3小时,反应完毕,静置(或离心)分离,澄清液收集后,有机锌试剂制备收率按100%计算,继续用于后续反应。
[0037]
实施例4氟伐他汀钠关键中间体(r=me)甲酯的合成
[0038][0039]
氮气保护下,向250ml反应瓶中投入0.5mol/l的brznch2co2me的四氢呋喃溶液100ml(50mmol),3.0ml的四甲基乙二胺(tmeda,20mmol),(e)-3-[3'-(4"
‑ꢀ
氟苯基)-1'-异丙基-1h-吲哚-2"-基]-2-丙烯醛3.1g(10mmol),搅拌混匀后升温至 50-60℃反应3小时,tlc法跟踪反应进程,反应完毕,降至室温,向反应液中缓慢滴加100ml水淬灭反应,乙酸乙酯萃取两次(100mlx2),合并有机相,依次用100ml的20%柠檬酸水溶液,100ml的5%碳酸氢钠水溶液,100ml水洗涤,有机相减压浓缩后用30ml正庚烷和15ml乙酸乙酯混合液重结晶得到淡棕色结晶产物2.59g,收率61.6%。
[0040]
实施例5氟伐他汀钠关键中间体(r=me)的合成
[0041]
氮气保护下,向250ml反应瓶中投入0.5mol/l的brznch2co2me的四氢呋喃溶液100ml(50mmol),0.30g碘化钠(2mmol),3.0ml的四甲基乙二胺(tmeda, 20mmol),(e)-3-[3'-(4"-氟苯基)-1'-异丙基-1h-吲哚-2"-基]-2-丙烯醛3.1g (10mmol),搅拌混匀后升温至50-60℃反应3小时,tlc法跟踪反应进程,反应完毕,降至室温,向反应液中缓慢滴加100ml水淬灭反应,乙酸乙酯萃取两次 (100mlx2),合并有机相,依次用100ml的20%柠檬酸水溶液,100ml的5%碳酸氢钠水溶液,100ml水洗涤,有机相减压浓缩后用30ml正庚烷和15ml乙酸乙酯混合液重结晶得到淡棕色结晶产物3.46g,收率82.4%。
[0042]
实施例6氟伐他汀钠关键中间体(r=me)的合成
[0043]
氮气保护下,向250ml反应瓶中投入0.5mol/l的brznch2co2me的四氢呋喃溶液100ml(50mmol),0.64g碘化锌(2mmol),3.0ml的四甲基乙二胺(tmeda, 20mmol),(e)-3-[3'-(4"-氟苯基)-1'-异丙基-1h-吲哚-2"-基]-2-丙烯醛3.1g (10mmol),搅拌混匀后升温至50-60℃反应3小时,tlc法跟踪反应进程,反应完毕,降至室温,向反应液中缓慢滴加100ml水淬灭反应,乙酸乙酯萃取两次 (100mlx2),合并有机相,依次用100ml的20%柠檬酸水溶液,100ml的5%碳酸氢钠水溶液,100ml水洗涤,有机相减压浓缩后用30ml正庚烷和15ml乙酸乙酯混合液重结晶得到淡棕色结晶产物3.86g,收率91.8%。
[0044]
实施例7氟伐他汀钠关键中间体(r=me)的合成
[0045]
氮气保护下,向250ml反应瓶中投入0.5mol/l的brznch2co2me的四氢呋喃溶液100ml(50mmol),0.38g碘化亚铜(2mmol),3.0ml的四甲基乙二胺 (tmeda,20mmol),(e)-3-[3'-(4"-氟苯基)-1'-异丙基-1h-吲哚-2"-基]-2-丙烯醛 3.1g(10mmol),搅拌混匀后升温至50-60℃反应3小时,tlc法跟踪反应进程,反应完毕,降至室温,向反应液中缓慢滴加100ml水淬灭反应,乙酸乙酯萃取两次(100mlx2),合并有机相,依次用100ml的20%柠檬酸水溶液,100ml的5%碳酸氢钠水溶液,100ml水洗涤,有机相减压浓缩后用30ml正庚烷和15ml乙酸乙酯混合液重结晶得到淡棕色结晶产物4.01g,收率95.5%。
[0046]
实施例8氟伐他汀钠关键中间体(r=me)的合成
[0047]
氮气保护下,向250ml反应瓶中投入0.5mol/l的brznch2co2me的四氢呋喃溶液100ml(50mmol),0.38g碘化亚铜(2mmol),3.12g 2,2'-联吡啶(2,2'-dipyridyl, 20mmol),(e)-3-[3'-(4"-氟苯基)-1'-异丙基-1h-吲哚-2"-基]-2-丙烯醛3.1g (10mmol),搅拌混匀后升温至50-60℃反应3小时,tlc法跟踪反应进程,反应完毕,降至室温,向反应液中缓慢滴加100ml水淬灭反应,乙酸乙酯萃取两次 (100mlx2),合并有机相,依次用100ml的20%柠檬酸水溶液,100ml的5%碳酸氢钠水溶液,100ml水洗涤,有机相减压浓缩后用30ml正庚烷和15ml乙酸乙酯混合液重结晶得到淡棕色结晶产物3.52g,收率83.7%。
[0048]
实施例9氟伐他汀钠关键中间体(r=me)的合成
[0049]
氮气保护下,向250ml反应瓶中投入0.5mol/l的brznch2co2me的四氢呋喃溶液100ml(50mmol),0.38g碘化亚铜(2mmol),1.96ml n,n
’‑
二甲基乙二胺(dmeda,20mmol),(e)-3-[3'-(4"-氟苯基)-1'-异丙基-1h-吲哚-2"-基]-2-丙烯醛3.1g(10mmol),搅拌混匀后升温至50-60℃反应3小时,tlc法跟踪反应进程,反应完毕,降至室温,向反应液中缓慢滴加100ml水淬灭反应,乙酸乙酯萃取两次(100mlx2),合并有机相,依次用100ml的20%柠檬酸水溶液,100ml 的5%碳酸氢钠水溶液,100ml水洗涤,有机相减压浓缩后用30ml正庚烷和15ml 乙酸乙酯混合液重结晶得到淡棕色结晶产物3.75g,收率89.2%。
[0050]
实施例10氟伐他汀钠关键中间体(r=et)的合成
[0051]
氮气保护下,向250ml反应瓶中投入0.5mol/l的brznch2co2et的四氢呋喃溶液100ml(50mmol),0.38g碘化亚铜(2mmol),3.0ml四甲基乙二胺(tmeda, 20mmol),(e)-3-[3'-(4"-氟苯基)-1'-异丙基-1h-吲哚-2"-基]-2-丙烯醛3.1g (10mmol),搅拌混匀后升温至50-60℃反应3小时,tlc法跟踪反应进程,反应完毕,降至室温,向反应液中缓慢滴加100ml水淬灭反应,乙酸乙酯萃取两次 (100mlx2),合并有机相,依次用100ml的20%柠檬酸水溶液,100ml的5%碳酸氢钠水溶液,100ml水洗涤,有机相减压浓缩后用30ml正庚烷和15ml乙酸乙酯混合液重结晶得到淡棕色结晶产物4.04g,收率92.3%。
[0052]
实施例11氟伐他汀钠关键中间体(r=i-pr)的合成
[0053]
氮气保护下,向250ml反应瓶中投入0.5mol/l的brznch2co
2-i-pr的四氢呋喃溶液100ml(50mmol),3.0ml的四甲基乙二胺(tmeda,20mmol),0.38g 碘化亚铜(2mmol),(e)-3-[3'-(4"-氟苯基)-1'-异丙基-1h-吲哚-2"-基]-2-丙烯醛3.1g (10mmol),搅拌混匀后升温至50-60℃反应3小时,tlc法跟踪反应进程,反应完毕,降至室温,向反应液中缓慢滴加100ml水淬灭反应,乙酸乙酯萃取两次 (100mlx2),合并有机相,依次用100ml的20%柠檬酸水溶液,100ml的5%碳酸氢钠水溶液,100ml水洗涤,有机相减压浓缩后用30ml正庚烷和15ml乙酸乙酯混合液重结晶得到淡棕色结晶产物4.10g,收率91.0%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1