氯沙坦-脂肪酸偶联前药化合物、纳米制剂及其制备方法和应用
1.本发明涉及肿瘤治疗药物技术领域,尤其涉及一种氯沙坦-脂肪酸偶联前药化合物、纳米制剂及其制备方法和应用。
背景技术:
2.纳米载药系统是肿瘤治疗领域的研究热点之一,由于其具有血液循环时间长、肿瘤组织选择性分布、肿瘤部位滞留时间长等优势,有望克服小分子化疗药物的局限性,达成高疗效和低毒性的平衡。
3.然而,部分肿瘤如胰腺癌以具有丰富且致密的细胞外基质为特征,严重限制了纳米载药系统的疗效。纤维化肿瘤基质压迫瘤内血管使血流灌注下降,直接抑制药物递送,同时基质本身作为物理屏障,阻挡纳米药物到达肿瘤细胞,降低了药物疗效。因此,寻找靶向肿瘤基质的治疗策略,是纳米药物递送领域亟待解决的问题。
4.氯沙坦是一种常用降压药。vikash p.chauhan等的研究提示,氯沙坦可抑制tgf-β通路的异常激活,从而抑制肿瘤细胞外基质纤维化进程(chauhan v p,martin j d,liu h,et al.nature communications[j]2013,4:2516)。在最近的一项ii期临床试验中,氯沙坦显著提高了接受folfirinox新辅助治疗的局部晚期胰腺癌患者的手术适用率,延长了患者的生存期。
[0005]
由于高血压是肿瘤患者的常见共病,而氯沙坦是临床上最常用的降压药之一,将氯沙坦应用于肿瘤基质调节治疗具有较大的临床转化价值。公开号为cn 110812356 a的中国专利文献公开了一种氯沙坦新抗肿瘤的应用,使用氯沙坦来抑制ang ii和ang ii 1型受体的结合,负责引发大部分与ras相关的活动,包括血管生成、细胞增殖和迁移,使得氯沙坦能够抑制肿瘤内部及肿瘤周边区域的血管生成、细胞增殖等活动,以直接和间接地双向抑制肿瘤细胞的增长(减少氧气和营养供给)。
[0006]
但氯沙坦具有较短的终末半衰期和较低的口服利用度,限制了其在肿瘤部位的蓄积和药效。因此,开发基于氯沙坦的纳米载药系统有望延长其体内循环时间,增加瘤内蓄积,提高肿瘤基质调节能力,并增强整体抗肿瘤疗效。
技术实现要素:
[0007]
本发明提供了一种氯沙坦-脂肪酸偶联前药化合物以及基于氯沙坦-脂肪酸偶联前药自组装而得到的纳米制剂,该纳米制剂可抑制肿瘤相关成纤维细胞激活,进而抑制肿瘤基质形成,增加肿瘤内的血管灌注,提高抗肿瘤药物在肿瘤组织的渗透性,从而增强抗肿瘤药物对肿瘤的治疗效果。
[0008]
本发明的技术方案如下:
[0009]
一种氯沙坦-脂肪酸偶联前药化合物,其结构如式(i)所示:
[0010][0011]
其中,r为饱和或不饱和脂肪酰基。
[0012]
优选的,r为正丁酰基、正癸酰基、油酰基、亚油酰基或亚麻酰基。
[0013]
所述的氯沙坦-脂肪酸偶联前药化合物的制备方法为:在缩合剂和催化剂作用下,将氯沙坦与r对应的脂肪酸进行酯化反应,得到所述的氯沙坦-脂肪酸偶联前药化合物。
[0014]
所述的缩合剂为1-(3-二甲氨基丙基)-3-乙基碳二亚胺(edc);所述的催化剂为4-二甲氨基吡啶(dmap)。
[0015]
优选的,氯沙坦-脂肪酸偶联前药化合物的制备方法中,溶剂为二氯甲烷(dcm)或氯仿。
[0016]
优选的,氯沙坦-脂肪酸偶联前药化合物的制备方法中,酯化反应温度为40~50℃;酯化反应时间为6~12h。
[0017]
优选的,所述的催化剂、缩合剂、脂肪酸与氯沙坦的摩尔比分别独立的为(1~2):(1~2):(0.7~1):1。
[0018]
本发明还提供了一种氯沙坦-脂肪酸偶联前药化合物的纳米制剂,所述的纳米制剂为氯沙坦-脂肪酸偶联前药化合物自组装纳米制剂或聚乙二醇化的氯沙坦-脂肪酸偶联前药化合物纳米制剂。
[0019]
所述的氯沙坦-脂肪酸偶联前药自组装纳米制剂的制备方法包括:将溶有氯沙坦-脂肪酸偶联前药化合物的有机溶剂于超声条件下注入水相中,透析得到均匀分散的纳米颗粒,即为氯沙坦-脂肪酸偶联前药自组装纳米制剂。
[0020]
所述的有机溶剂为有机溶剂为二甲基亚砜;二甲基亚砜与水相体积比为1:8~15;进一步优选的,所述体积比1:9。
[0021]
所述的聚乙二醇化的氯沙坦-脂肪酸偶联前药化合物纳米制剂的制备方法包括:将溶有氯沙坦-脂肪酸偶联前药化合物与1,2-二硬脂酰基-sn-甘油-3-磷酸乙醇胺-n-[甲氧基(聚乙二醇)](dspe-peg)的有机溶剂于超声条件下注入水相中,透析得到均匀分散的纳米颗粒,即为聚乙二醇化氯沙坦-脂肪酸偶联前药化合物纳米制剂。
[0022]
优选的,氯沙坦-脂肪酸偶联前药化合物与1,2-二硬脂酰基-sn-甘油-3-磷酸乙醇胺-n-[甲氧基(聚乙二醇)]的质量比为8~12:1,最优选为10:1。
[0023]
优选的,所述的1,2-二硬脂酰基-sn-甘油-3-磷酸乙醇胺-n-[甲氧基(聚乙二醇)]为dspe-peg
2k
(dspe-peg
2000
)。
[0024]
制备过程中,可将氯沙坦-脂肪酸偶联前药化合物、dspe-peg
2k
分别溶于有机溶剂中,再将有机溶剂充分混匀,最后将混匀的有机溶剂于超声条件下注入水相中,透析得到均匀分散的纳米颗粒。
[0025]
优选的,所述的有机溶剂为二甲基亚砜;二甲基亚砜与水相体积比为1:8~15;进
一步优选的,所述体积比为1:9。
[0026]
本发明还提供了氯沙坦-脂肪酸偶联前药化合物的纳米制剂在制备调节肿瘤基质的药物中的应用。
[0027]
本发明还提供了氯沙坦-脂肪酸偶联前药化合物的纳米制剂与7-乙基-10-羟基喜树碱(sn38)联合使用在制备抗肿瘤纳米药物中的应用。
[0028]
优选的,所述的氯沙坦-脂肪酸偶联前药化合物的纳米制剂为聚乙二醇化氯沙坦-脂肪酸偶联前药化合物纳米制剂。
[0029]
小鼠肿瘤实验结果表明,相较于游离型氯沙坦,聚乙二醇化氯沙坦-脂肪酸偶联前药化合物纳米制剂体现出对抗肿瘤纳米药物更强的疗效增强作用。同样与基于sn38的抗肿瘤纳米药物联合使用,与生理盐水对照组相比,氯沙坦纳米制剂组的肿瘤缩小了4.4倍;与同等剂量下的游离型氯沙坦组相比,氯沙坦纳米制剂组的肿瘤缩小了2.7倍。
[0030]
与现有技术相比,本发明的优势效果体现在:
[0031]
1)本发明对肿瘤基质调节药物的选择充分考虑临床应用,氯沙坦是最常用的抗高血压药物之一,其安全性已得到广泛验证,用于修饰氯沙坦的脂肪酸为人体所需物质或天然产物,生物相容性好,同时氯沙坦-脂肪酸前药制备步骤简单,成本较低,具有较大的临床转化潜力;
[0032]
2)本发明构建的氯沙坦-脂肪酸纳米制剂由小分子前药自组装得到,避免了由载体材料带来的安全性问题,有利于临床转化;
[0033]
3)本发明构建的氯沙坦-脂肪酸纳米制剂显著改善了氯沙坦的药代动力学特性,延长其体内循环时间,同时可以通过epr效应(增强渗透滞留效应)实现肿瘤被动靶向效果,从而发挥更强的肿瘤基质调节作用,促进抗肿瘤药物向肿瘤组织渗透,增强整体疗效。
附图说明
[0034]
图1-5为实施例1-5中氯沙坦-脂肪酸偶联前药的核磁图谱;
[0035]
图6-11为实施例6-11中氯沙坦纳米制剂的粒径分布图;
[0036]
图12-17为实施例6-11中氯沙坦纳米制剂的透射电镜图;
[0037]
图18为实施例11中氯沙坦纳米制剂的体外释放;图中,los nb为氯沙坦纳米制剂,los@dspe-peg
2k micelles为dspe-peg
2k
搭载的游离氯沙坦;
[0038]
图19为实施例11中氯沙坦纳米制剂对瘤内肿瘤相关成纤维细胞激活情况的影响;图中,saline为生理盐水对照,free los为游离氯沙坦,los nb为氯沙坦纳米制剂;
[0039]
图20为实施例11中氯沙坦纳米制剂对肿瘤胶原浓度的影响;图中,saline为生理盐水对照,free los为游离氯沙坦,los nb为氯沙坦纳米制剂;
[0040]
图21为实施例11中氯沙坦纳米制剂对瘤内血流灌注的影响;图中,saline为生理盐水对照,free los为游离氯沙坦,los nb为氯沙坦纳米制剂;
[0041]
图22为实施例11中氯沙坦纳米制剂对后续纳米药物瘤内分布的影响;图中,saline为生理盐水对照,free los为游离氯沙坦,los nb为氯沙坦纳米制剂;
[0042]
图23为实施例11中氯沙坦纳米制剂对后续纳米药物治疗效果的影响;图中,saline为生理盐水对照,free los为游离氯沙坦,los nb为氯沙坦纳米制剂;sn38 np为sn38纳米药物;free los+sn38 np为游离氯沙坦与sn38纳米药物联用;los nb+sn38 np为
氯沙坦纳米制剂与sn38纳米药物联用。
具体实施方式
[0043]
实施例1
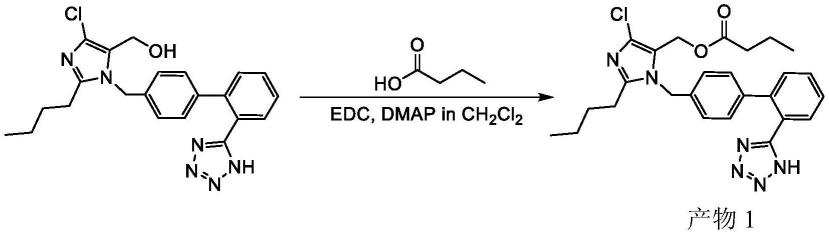
[0044]
氯沙坦-正丁酸偶联前药的合成,如下所示:
[0045][0046]
在反应瓶中依次加入氯沙坦(100.0mg,0.236mmol)、正丁酸(20.8mg,0.236mmol)和dmap(43.3mg,0.354mmol),溶解于3ml无水dcm,再快速滴加edc(90.7mg,0.472mmol)。70℃搅拌12小时,利用薄层色谱观察反应情况。当反应基本结束时,冷却反应液,用饱和食盐水清洗;有机相用无水硫酸钠干燥,过滤,滤液旋蒸除去溶剂。通过柱层析色谱分离纯化(dcm:meoh=80:1)后得到产物1(47.1mg,产率40.4%)。
[0047]
产物1的1h nmr核磁数据如下,核磁图谱如图1所示:
[0048]1h nmr(400mhz,chloroform-d)δ7.86(d,j=7.6hz,1h),7.55(dt,j=31.0,7.6hz,2h),7.38(d,j=7.6hz,1h),7.03(d,j=7.7hz,2h),6.77(d,j=7.8hz,2h),5.09(s,2h),4.78(s,2h),2.46(t,j=7.8hz,2h),2.11(t,j=7.5hz,2h),1.61(t,j=7.8hz,2h),1.51(q,j=7.4hz,2h),1.32(d,j=7.8hz,2h),0.89
–
0.83(m,6h).
[0049]
实施例2
[0050]
氯沙坦-正癸酸偶联前药的合成,如下所示:
[0051][0052]
在反应瓶中依次加入氯沙坦(100.0mg,0.236mmol)、正癸酸(40.7mg,0.236mmol)和dmap(43.3mg,0.354mmol),溶解于3ml无水dcm,再快速滴加edc(90.7mg,0.472mmol)。70℃搅拌12小时,利用薄层色谱观察反应情况。当反应基本结束时,冷却反应液,用饱和食盐水清洗;有机相用无水硫酸钠干燥,过滤,滤液旋蒸除去溶剂。通过柱层析色谱分离纯化(dcm:meoh=80:1)后得到产物2(52.7mg,产率38.6%)。
[0053]
产物2的1h nmr核磁数据如下,核磁图谱如图2所示:
[0054]1h nmr(400mhz,chloroform-d)δ7.90(d,j=7.6hz,1h),7.57(dt,j=26.3,7.5hz,2h),7.41(d,j=7.6hz,1h),7.10(d,j=7.8hz,2h),6.80(d,j=7.8hz,2h),5.13(s,2h),4.83(s,2h),2.42(t,j=7.8hz,2h),2.09(t,j=7.6hz,2h),1.59(t,j=7.8hz,2h),
1.46(q,j=7.4hz,2h),1.37
–
1.19(m,14h),0.87(td,j=7.2,3.2hz,6h).
[0055]
实施例3
[0056]
氯沙坦-油酸偶联前药的合成,如下所示:
[0057][0058]
在反应瓶中依次加入氯沙坦(100.0mg,0.236mmol)、油酸(66.8mg,0.236mmol)和dmap(43.3mg,0.354mmol),溶解于3ml无水dcm,再快速滴加edc(90.7mg,0.472mmol)。70℃搅拌12小时,利用薄层色谱观察反应情况。当反应基本结束时,冷却反应液,用饱和食盐水清洗;有机相用无水硫酸钠干燥,过滤,滤液旋蒸除去溶剂。通过柱层析色谱分离纯化(dcm:meoh=80:1)后得到产物3(68.2mg,产率42.0%)。
[0059]
产物3的1h nmr核磁数据如下,核磁图谱如图3所示:
[0060]1h nmr(400mhz,chloroform-d)δ7.96(d,j=7.6hz,1h),7.57(dt,j=23.7,7.6hz,2h),7.41(d,j=7.5hz,1h),7.12(d,j=7.8hz,2h),6.83(d,j=7.8hz,2h),5.32(q,j=6.4hz,2h),5.14(s,2h),4.83(s,2h),2.45(t,j=7.9hz,2h),2.10(t,j=7.6hz,2h),2.02
–
1.95(m,4h),1.62(q,j=7.7hz,2h),1.47(p,j=7.1hz,2h),1.36
–
1.22(m,22h),0.88(td,j=7.2,3.1hz,6h).
[0061]
实施例4
[0062]
氯沙坦-油酸偶联前药的合成,如下所示:
[0063][0064]
在反应瓶中依次加入氯沙坦(100.0mg,0.236mmol)、亚油酸(66.3mg,0.236mmol)和dmap(43.3mg,0.354mmol),溶解于3ml无水dcm,再快速滴加edc(90.7mg,0.472mmol)。70℃搅拌12小时,利用薄层色谱观察反应情况。当反应基本结束时,冷却反应液,用饱和食盐水清洗;有机相用无水硫酸钠干燥,过滤,滤液旋蒸除去溶剂。通过柱层析色谱分离纯化(dcm:meoh=80:1)后得到产物4(70.1mg,产率47.3%)。
[0065]
产物4的1h nmr核磁数据如下,核磁图谱如图4所示:
[0066]1h nmr(400mhz,chloroform-d)δ7.93(dd,j=7.6,1.5hz,1h),7.58(dtd,j=24.2,7.6,1.5hz,2h),7.41(dd,j=7.5,1.4hz,1h),7.16
–
7.08(m,2h),6.80(d,j=8.0hz,2h),5.40
–
5.30(m,4h),5.14(s,2h),4.84(s,2h),2.75(t,j=6.2hz,2h),2.44
–
2.36(m,2h),2.11
–
1.98(m,6h),1.62
–
1.54(m,2h),1.45(q,j=7.3hz,2h),1.36
–
1.18(m,16h),0.88(td,j=7.1,2.7hz,6h).
[0067]
实施例5
[0068]
氯沙坦-油酸偶联前药的合成,如下所示:
[0069][0070]
在反应瓶中依次加入氯沙坦(100.0mg,0.236mmol)、亚麻酸(65.8mg,0.236mmol)和dmap(43.3mg,0.354mmol),溶解于3ml无水dcm,再快速滴加edc(90.7mg,0.472mmol)。70℃搅拌12小时,利用薄层色谱观察反应情况。当反应基本结束时,冷却反应液,用饱和食盐水清洗;有机相用无水硫酸钠干燥,过滤,滤液旋蒸除去溶剂。通过柱层析色谱分离纯化(dcm:meoh=80:1)后得到产物5(65.5mg,产率40.5%)。
[0071]
产物5的1h nmr核磁数据如下,核磁图谱如图5所示:
[0072]1h nmr(400mhz,chloroform-d)δ7.94(d,j=7.6hz,1h),7.57(dt,j=24.4,7.5hz,2h),7.40(d,j=7.5hz,1h),7.11(d,j=7.8hz,2h),6.83(d,j=7.8hz,2h),5.41
–
5.29(m,6h),5.13(s,2h),4.83(s,2h),2.79(q,j=5.3,4.9hz,4h),2.45(t,j=7.9hz,2h),2.06(dt,j=23.4,7.4hz,6h),1.61(t,j=7.8hz,2h),1.47(t,j=7.3hz,2h),1.39
–
1.22(m,10h),0.97(t,j=7.5hz,3h),0.88(t,j=7.3hz,3h).
[0073]
实施例6
[0074]
氯沙坦-正丁酸自组装纳米制剂的制备
[0075]
制备氯沙坦最终浓度为1mg/ml的氯沙坦-正丁酸自组装纳米制剂。将1.166mg的氯沙坦-正丁酸(实施例1制备得到)溶于100ul二甲基亚砜溶液,于超声条件下缓慢注入900μl的超纯水中,使用透析法(截留分子量3500)除去残留的dmso,得到均匀分散的氯沙坦-正丁酸自组装纳米制剂。其粒径分布和透射电镜如图6、12所示。
[0076]
实施例7
[0077]
氯沙坦-正癸酸自组装纳米制剂的制备
[0078]
制备氯沙坦最终浓度为1mg/ml的氯沙坦-正癸酸自组装纳米制剂。将1.365mg的氯沙坦-正癸酸(实施例2制备得到)溶于100ul二甲基亚砜溶液,于超声条件下缓慢注入900μl的超纯水中,使用透析法(截留分子量3500)除去残留的dmso,得到均匀分散的氯沙坦-正癸酸自组装纳米制剂。其粒径分布和透射电镜如图7、13所示。
[0079]
实施例8
[0080]
氯沙坦-油酸自组装纳米制剂的制备
[0081]
制备氯沙坦最终浓度为1mg/ml的氯沙坦-油酸自组装纳米制剂。将1.625mg的氯沙坦-油酸(实施例3制备得到)溶于100ul二甲基亚砜溶液,于超声条件下缓慢注入900μl的超纯水中,使用透析法(截留分子量3500)除去残留的dmso,得到均匀分散的氯沙坦-油酸自组装纳米制剂。其粒径分布和透射电镜如图8、14所示。
[0082]
实施例9
[0083]
氯沙坦-亚油酸自组装纳米制剂的制备
[0084]
制备氯沙坦最终浓度为1mg/ml的氯沙坦-亚油酸自组装纳米制剂。将1.621mg的氯沙坦-亚油酸(实施例4制备得到)溶于100ul二甲基亚砜溶液,于超声条件下缓慢注入900μl的超纯水中,使用透析法(截留分子量3500)除去残留的dmso,得到均匀分散的氯沙坦-亚油
酸自组装纳米制剂。其粒径分布和透射电镜如图9、15所示。
[0085]
实施例10
[0086]
氯沙坦-亚麻酸自组装纳米制剂的制备
[0087]
制备氯沙坦最终浓度为1mg/ml的氯沙坦-亚麻酸自组装纳米制剂。将1.616mg的氯沙坦-亚麻酸(实施例5制备得到)溶于100ul二甲基亚砜溶液,于超声条件下缓慢注入900μl的超纯水中,使用透析法(截留分子量3500)除去残留的dmso,得到均匀分散的氯沙坦-亚麻酸自组装纳米制剂。其粒径分布和透射电镜如图10、16所示。
[0088]
实施例11
[0089]
聚乙二醇化氯沙坦-亚油酸纳米制剂的制备
[0090]
制备氯沙坦最终浓度为1mg/ml的聚乙二醇化氯沙坦-亚油酸纳米制剂。将1.621mg的氯沙坦-亚油酸(实施例4制备得到)与0.162g dspe-peg
2000
(氯沙坦-亚油酸与dspe-peg
2000
的质量比为10:1)溶于100ul二甲基亚砜溶液,于超声条件下缓慢注入900μl的超纯水中,使用透析法(截留分子量3500)除去残留的dmso,得到均匀分散的聚乙二醇化氯沙坦-亚油酸纳米制剂。其粒径分布如图11所示,可见水和动力学直径约为59.1nm,呈单一峰分布,说明该纳米制剂粒径分布均一。其透射电镜如图17所示,直观地显示出规整的球形形貌、均一的粒径和高度的分散性。
[0091]
实施例12
[0092]
聚乙二醇化氯沙坦-亚油酸纳米制剂的体外释放
[0093]
将实施例11中制备的纳米制剂3ml置于分子量为7000的透析袋中,置于外界20ml ph为7.4的磷酸缓冲液中,在温度为37℃,转速为150r/min的环境中,分别在24h、72h、120h、168h取出外界磷酸缓冲液,通过高效液相检测得到纳米制剂的体外释放情况。由图18可知,与单纯使用dspe-peg
2000
包裹的游离氯沙坦相比,基于氯沙坦-亚油酸偶联前药自组装的聚乙二醇化纳米制剂具有明显的缓释特性,有利于其在体内长时间循环。
[0094]
实施例13
[0095]
聚乙二醇化氯沙坦-亚油酸纳米制剂使用后对肿瘤基质形成的影响
[0096]
本发明采用panc02鼠源胰腺癌原位移植小鼠模型对实施例11中制备的纳米制剂进行肿瘤基质调节能力评价。将luciferase稳转的鼠源胰腺癌panc02细胞系按2
×
105个细胞注射到小鼠胰尾,建立原位胰腺癌小鼠模型。待肿瘤细胞注射1周后将荷瘤小鼠随机分为3组,每组5只。实验组通过尾静脉注射分别给予氯沙坦纳米制剂(由实施例11中制备得到,氯沙坦等效剂量为20mg/kg/d,采用los nb表示)以及同等剂量的游离氯沙坦(采用free los表示),对照组注射相同体积的生理盐水(采用saline表示)。每天注射1次,共注射7次。最后一次给药结束24小时后,收集小鼠肿瘤组织。采用免疫荧光标记α-平滑肌肌动蛋白(α-sma),作为激活的胰腺癌相关成纤维细胞的标志物。免疫荧光图片及其半定量结果如图19所示,氯沙坦纳米制剂组的小鼠肿瘤组织α-sma表达水平显著低于游离氯沙坦组和对照组,表明氯沙坦纳米制剂可以有效抑制胰腺癌相关成纤维细胞的激活。取部分小鼠肿瘤组织研磨成匀浆,通过sircol胶原检测试剂盒对肿瘤胶原进行定量,评价肿瘤基质的形成水平。结果如图20所示,氯沙坦纳米制剂组的小鼠肿瘤组织胶原浓度显著低于游离氯沙坦组和对照组,表明氯沙坦纳米制剂可以有效减少肿瘤细胞外基质的形成。
[0097]
实施例14
[0098]
聚乙二醇化氯沙坦-亚油酸纳米制剂使用后对瘤内血流灌注的影响
[0099]
小鼠模型、分组及给药方式同实施例13。最后一次给药结束24小时后,尾静脉注射给予488标记的凝集素(488-lectin),剂量为5mg/kg,1小时后收集小鼠肿瘤组织,制备冰冻切片,采用cd31抗体对血管内皮细胞进行免疫荧光染色后,分析功能化血管(lectin
+
cd31
+
)占所有血管(cd31
+
)的比例,评价肿瘤组织内的血管灌注情况。结果如图21所示,氯沙坦纳米制剂组的小鼠肿瘤组织内部功能化血管显著多于游离氯沙坦组和对照组,表明氯沙坦纳米制剂可以有效提升肿瘤内的血管灌注。
[0100]
实施例15
[0101]
聚乙二醇化氯沙坦-亚油酸纳米制剂使用后对后续纳米药物瘤内分布的影响
[0102]
小鼠模型、分组及给药方式同实施例13。最后一次给药结束24小时后,尾静脉注射用近红外染料dir标记的纳米粒(dir剂量为10μg/kg)。在注射后0.5h、2h、4h、6h、8h、12h、24h通过活体成像观察dir标记的纳米粒在小鼠体内的分布。24小时后收集小鼠肿瘤组织及其他主要器官(包括心、肺、肝、脾、肾),再次通过活体成像分析离体组织中的荧光分布。结果如图22所示,氯沙坦纳米制剂处理更利于后续纳米药物在肿瘤部位的聚集。离体组织成像结果证实,氯沙坦纳米制剂组的小鼠肿瘤组织荧光强度显著高于游离氯沙坦组和对照组,表明氯沙坦纳米制剂可以有效增加后续纳米药物在肿瘤组织内的分布。
[0103]
实施例16
[0104]
聚乙二醇化氯沙坦-亚油酸纳米制剂使用后对后续纳米药物治疗胰腺癌效果的影响
[0105]
小鼠模型同实施例13。以注射肿瘤细胞记为第0天,将荷瘤小鼠随机分为6组,每组5只。对照组(同步其他组给药时间,尾静脉注射给予生理盐水)、游离氯沙坦组(第6-12天每天尾静脉注射给予游离氯沙坦,第13、16、19天尾静脉给予生理盐水)、氯沙坦纳米制剂组(第6-12天每天尾静脉注射给予氯沙坦纳米制剂,第13、16、19天尾静脉给予生理盐水)、sn38纳米药物组(第6-12天每天尾静脉注射给予生理盐水,第13、16、19天尾静脉给予sn38纳米药物)、游离氯沙坦-sn38纳米药物联用组(第6-12天每天尾静脉注射给予游离氯沙坦,第13、16、19天尾静脉给予sn38纳米药物)、氯沙坦纳米制剂-sn38纳米药物联用组(第6-12天每天尾静脉注射给予氯沙坦纳米制剂,第13、16、19天尾静脉给予sn38纳米药物)。游离氯沙坦及氯沙坦纳米制剂剂量均为等效氯沙坦20mg/kg/d,sn38纳米药物剂量为8mg/kg/d。第12、16、20、26天通过活体成像评估小鼠原位肿瘤大小。结果如图23所示,按活体成像肿瘤信号抑制率和肿瘤重量抑制率计算,sn38纳米药物组对肿瘤的抑制率分别为41.5%和76.1%,游离氯沙坦-sn38纳米药物联用组对肿瘤的抑制率分别为63.7%和82.8%,而氯沙坦纳米制剂-sn38纳米药物联用组对肿瘤的抑制率分别为86.7%和96.5%。上述结果说明氯沙坦纳米制剂的处理可以有效增强纳米药物对肿瘤的治疗效果,且其增强能力显著优于游离氯沙坦。
[0106]
以上所述的实施例对本发明的技术方案和有益效果进行了详细说明,应理解的是以上所述仅为本发明的具体实施例,并不用于限制本发明,凡在本发明的原则范围内所做的任何修改、补充和等同替换等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1