一种头孢克肟的合成方法与流程
1.本发明涉及一种头孢克肟的合成方法,具体涉及一种利用新型的活性中间体实现7-avna和氨基噻唑乙酰肟酸的缩合方法,属于药物合成技术领域。
背景技术:
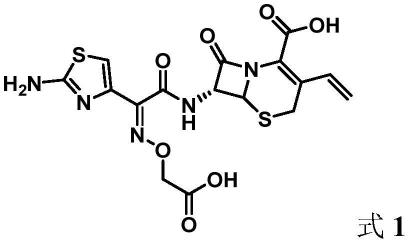
2.头孢克肟(英文名:cefixime),化学名:7-[(z)-2-(2-氨基-4-噻唑基)-2-(羧甲氧亚胺基)乙酰氨基]-8-氧代-3-乙烯-5-硫杂-1-氮杂二环[4,2,0]-辛-2-烯-2-羧酸。头孢克肟的分子量:453.45;cas登记号:79350-37-1;结构式如式1所示:
[0003][0004]
头孢克肟属于新型第三代高效广谱的头孢类抗菌素,是临床治疗中主要应用的抗生素品种。适用于治疗敏感菌所致的呼吸、泌尿和胆道等部位的感染。头孢克肟对革兰阴性杆菌产生的β-内酰胺酶高度稳定,对革兰阴性杆菌抗菌作用强于第一代和第二代头孢菌素,对革兰阳性球菌抗菌作用不如第一代和第二代头孢菌素。
[0005]
现有技术中,合成头孢克肟的路线中,侧链的引入主要采用的是7-avna与相应的mica活化硫酯或地尼活化硫酯反应获得(1:j.am.chem.soc.1955,77,1067-1068.2:j.am.chem.soc.1969,91,5669-5671.3:j.org.chem.2001,66,5245-5247.4:tetrahedron2004,60,2447-2467.5:tetrahedron2005,61,10827-10852.6:angew.chem.,int.ed.2008,47,10030-10074.7:chem.soc.rev.2009,38,606-631.8:nature.2011,480,471-479.9:chem.rev.2011,111,6557-6602.10:j.am.chem.soc.2016,138,13135
–
13138.)。而mica活化硫酯或地尼活化硫酯需要通过相应的羧酸在有机磷的作用下与硫试剂合成。整个侧链至少需要两步引入,涉及两步分离操作,反应收率不高,试剂昂贵,副产物硫化物不易分离,对环境影响大。
[0006]
技术实现要素:
[0007]
为了克服现有技术中存在的不足,本发明提供了一种新的头孢克肟的合成方法,该方法利用了新型的活性中间体实现7-avna和氨基噻唑乙酰肟酸的缩合,缩短了操作步骤,提高了反应收率,减少了副产物。
[0008]
为解决上述技术问题,本发明提供了以下技术方案:
[0009]
一种头孢克肟的合成方法,包括:
[0010]
(1)化合物(2)与n-乙炔基-n,4-二甲基苯磺酰胺(3)进行加成反应得到中间体(4);
[0011]
(2)中间体(4)与7-avna再进行亲核取代反应得到头孢克肟;
[0012]
反应式如下:
[0013][0014]
本发明中,采用n-乙炔基-n,4-二甲基苯磺酰胺(3)作为加成反应的试剂,与化合物(2)反应得到新的活性中间体(该活性中间体的制备方法可参考:1.chemical communications(cambridge,united kingdom)(2021),57(55),6792-6795),然后再与7-avna进行亲核取代反应,可以高收率地得到头孢克肟,减少副产物的生成,并且不采用有机磷试剂,对环境更加友好。
[0015]
作为优选,所述合成方法为一锅法,步骤(1)的反应结束之后得到中间体(4)不进行纯化,直接向反应液中加入7-avna进行步骤(2)的反应。由于步骤(1)的反应转化率高,反应结束之后不需要经过额外的提纯,有效缩短了操作步骤,更加适合工业化生产。
[0016]
作为优选,步骤(1)和步骤(2)的反应在有机溶剂中进行,所述的有机溶剂为醚类溶剂、酯类溶剂、卤代烃类溶剂或芳烃类溶剂中的一种或几种;作为进一步的优选,所述有机溶剂为四氢呋喃,甲苯,1,2-二氯乙烷,二氯甲烷,乙酸乙酯中的一种或几种,作为最优选,所述有机溶剂为二氯甲烷。
[0017]
作为进一步优选,所述的有机溶剂只在步骤(1)中加入,步骤(2)不加入所述有机溶剂;
[0018]
所述的化合物(2)与所述有机溶剂的用量比为1mmol:3~7ml。
[0019]
作为优选,所述加成反应和亲核取代反应的温度可以相同,也可以不痛,温度范围为0-50℃,进一步优选为20~35℃,最优选为25℃。该反应温度条件温和,并且反应收率高。
[0020]
作为优选,所述化合物(2)、n-乙炔基-n,4-二甲基苯磺酰胺(3)和7-avna的摩尔比例为1.0:1.0-1.5:1.0-1.5,进一步优选为1.0:1.0:1.2。
[0021]
本发明中,步骤(1)和步骤(2)的反应时间无特别的限定,一般通过tlc或者hplc监测到原料反应完全即可停止。
[0022]
同现有技术相比,本发明的有益效果体现在:
[0023]
本发明在温和条件下实现了一锅缩合制备头孢克肟,缩短了反应步骤,提高了产
品收率和质量,所采用的试剂便宜易得,副产物容易分离,降低了能耗和污染。
具体实施方式
[0024]
下面结合实例对本发明作更进一步的说明。
[0025]
实施例1化合物头孢克肟(1)的制备
[0026]
在室温(25℃)条件下,往250ml圆底烧瓶加入4.90g(20mmol)氨基噻唑乙酰肟酸类似物(2)和4.20g(20mmol)化合物(3),加入100ml二氯甲烷搅拌,至到化合物(3)完全反应完。将化合物7-avna 5.00g(22mmol)加入到上述溶液中继续在室温和空气中搅拌直至中间体(4)完全反应完。将反应混合物浓缩并通过硅胶色谱柱纯化,以94.0%的产率得到头孢克肟(1)8.52g,hplc纯度为99.4%。
[0027]1h nmr(600mhz,methanol-d4)δ:7.12(q,j=28.9hz,1h),6.96(s,1h),5.56(d,j=18.0hz,1h),5.88(d,j=4.7hz,1h),5.32(d,j=11.4hz,1h),5.21(d,j=4.8hz,1h),4.75(s,2h),3.78(d,j=17.5hz,1h),3.61(d,j=17.4hz,1h).
13
c nmr(600mhz,methanol-d4)δ:174.0,171.5,165.3,164.2,150.2,141.2,133.4,127.6,126.8,117.6,113.1,71.9,60.5,59.2,24.9.
[0028]
实施例2化合物头孢克肟(1)的制备
[0029]
在室温(25℃)条件下,往250ml圆底烧瓶加入4.90g(20mmol)氨基噻唑乙酰肟酸类似物(2)和4.20g(20mmol)化合物(3),加入100ml四氢呋喃搅拌,至到化合物(3)完全反应完。将化合物7-avna 5.00g(22mmol)加入到上述溶液中继续在室温和空气中搅拌直至中间体(4)完全反应完。将反应混合物浓缩并通过硅胶色谱柱纯化,以83.4%的产率得到头孢克肟(1)7.56g,hplc纯度为99.0%。
[0030]
实施例3化合物头孢克肟(1)的制备
[0031]
在室温(25℃)条件下,往250ml圆底烧瓶加入4.90g(20mmol)氨基噻唑乙酰肟酸类似物(2)和4.20g(20mmol)化合物(3),加入100ml甲苯搅拌,至到化合物(3)完全反应完。将化合物7-avna 5.00g(22mmol)加入到上述溶液中继续在室温和空气中搅拌直至中间体(4)完全反应完。将反应混合物浓缩并通过硅胶色谱柱纯化,以86.8%的产率得到头孢克肟(1)7.87g,hplc纯度为98.9%。
[0032]
实施例4化合物头孢克肟(1)的制备
[0033]
在室温(25℃)条件下,往250ml圆底烧瓶加入4.90g(20mmol)氨基噻唑乙酰肟酸类似物(2)和4.20g(20mmol)化合物(3),加入100ml乙酸乙酯搅拌,至到化合物(3)完全反应完。将化合物7-avna 5.00g(22mmol)加入到上述溶液中继续在室温和空气中搅拌直至中间体(4)完全反应完。将反应混合物浓缩并通过硅胶色谱柱纯化,以89.7%的产率得到头孢克肟(1)8.13g,hplc纯度为98.6%。
[0034]
实施例5
[0035]
在35℃条件下,往250ml圆底烧瓶加入4.90g(20mmol)氨基噻唑乙酰肟酸类似物(2)和4.20g(20mmol)化合物(3),加入100ml二氯甲烷搅拌,至到化合物(3)完全反应完。将化合物7-avna 5.00g(22mmol)加入到上述溶液中继续在室温和空气中搅拌直至中间体(4)完全反应完。将反应混合物浓缩并通过硅胶色谱柱纯化,以91.2%的产率得到头孢克肟(1)8.27g,hplc纯度为98.8%。
[0036]
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书内容所做的等效结构或等效流程变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明专利保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1