用于检测伴侣基因的方法与流程
用于检测伴侣基因的方法
1.本技术是申请日为2020年08月31日,申请号为cn202010895988.8,发明名称为用于检测基因融合的靶向测序方法的分案申请。
技术领域
2.本发明涉及靶向方法,更具体地,本发明涉及用于检测伴侣基因的方法。
背景技术:
3.通过染色体重排,例如异位、缺失、插入,癌细胞常常发生基因融合事件。随着对融合基因的临床重要性的认识日益增加,融合基因的精确诊断也越来越受到重视。目前,已有多种抑制融合基因的肿瘤靶向治疗药物,包括伊马替尼/bcr-abl1、克唑替尼/eml4-alk和拉罗替尼/ntrk融合等。
4.融合基因的快速、准确诊断不但可以对癌症进行诊断和分型,还能够为后续的治疗提供必要信息。目前,融合基因诊断主要包括荧光原位杂交(fish)、ihc等方法,然而,这些检测方法的通量通常较低,依赖检验人员的经验,只适用于已知的融合亚型。有报道表明,使用ihc检测ntrk3融合的灵敏度降低,只有79%[identifying patients with ntrk fusion cancer.]。
[0005]
多种新兴的融合基因检测技术虽然避免了技术人员的主观判断,具有更高的通量和检测灵敏度,例如nanostring(ncounter vantage 3dtm assays)和agena massarray,但它们都不能发现新的融合亚型[overview of fusion detection strategies using next-generation sequencing]。此外,基于下一代测序的全转录组测序成本较高,且不能兼容临床样本、而多重扩增子靶向测序技术假阳性率较高、需要事先建立人群基线,利用生物信息学方法过滤低置信度的结果。
技术实现要素:
[0006]
针对现有技术中的至少部分技术问题,本发明提供一种用于检测基因融合的靶向测序方法。与传统探针设计相比,本发明的探针与目标外显子区域具有更高的结合效率,同时探针数量更少,覆盖区间更小,成本更低。
[0007]
本发明的用于检测基因融合的靶向测序方法,其包括以下步骤:
[0008]
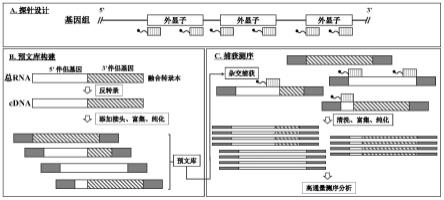
(1)预文库构建,其包括提取样本的总rna,反转录得到cdna,然后片段化为150-250pb的片段、并在末端修复、加a,连接测序接头,得到预文库;
[0009]
(2)利用封闭阻断剂封闭所述预文库,然后使探针组与所述预文库杂交,获得捕获片段,经pcr富集和纯化后质检,得到测序文库;
[0010]
(3)利用二代测序对所述测序文库进行测序,获得基因融合信息;
[0011]
其中,所述探针组由能够与基因a各外显子的5’末端和/或3’末端互补的第一探针或能够与基因b各外显子的5’末端和/或3’末端互补的第二探针组成。
[0012]
根据本发明所述的靶向测序方法,优选地,5’末端是指外显子5’端第1个碱基至第
120个碱基之间的片段,3’末端是指外显子3’端最后1个碱基至倒数第120个碱基之间的片段。
[0013]
根据本发明所述的靶向测序方法,优选地,所述基因a为5’伴侣基因,且所述第一探针与所述5’伴侣基因的各外显子的3’末端互补;所述基因b为3’伴侣基因,且所述第二探针与所述3’伴侣基因的5’末端互补。
[0014]
根据本发明所述的靶向测序方法,优选地,所述第一探针和所述第二探针分别为5’端生物素修饰的探针。
[0015]
根据本发明所述的靶向测序方法,优选地,所述步骤(1)为利用总rna进行文库构建,并不去除核糖体rna。
[0016]
根据本发明所述的靶向测序方法,优选地,所述步骤(1)中,在连接测序接头后进一步包括pcr富集和纯化步骤。
[0017]
根据本发明所述的靶向测序方法,优选地,步骤(2)的杂交步骤中,所述预文库为多个不同的预文库。
[0018]
本发明通过优化探针设计,减少了探针数量,并同时通过优化杂交和探针标记物结构提高捕获效果,从而使本发明在使用更少的探针和更低的测序数据量时,获得比传统捕获效率更高更多的阳性reads,减少了探针合成和测序成本,大大提高了检测灵敏度。此外,不需要融合基因的先验知识,能够对未知融合亚型进行检测。
附图说明
[0019]
图1为本发明示例性融合基因检测的流程图。
[0020]
图2为本发明的一种示例性探针设计方法与传统方法的比较示意图。
[0021]
图3为探针与目的片段不同结合长度对结合效率的影响。
[0022]
图4为本发明对alk转录本进行探针设计的示意图。
[0023]
图5为本发明设计的eml:exon13/alk:exon20融合探针的示意图。
具体实施方式
[0024]
现详细说明本发明的多种示例性实施方式,该详细说明不应认为是对本发明的限制,而应理解为是对本发明的某些方面、特性和实施方案的更详细的描述。
[0025]
应理解本发明中所述的术语仅仅是为描述特别的实施方式,并非用于限制本发明。另外,对于本发明中的数值范围,应理解为具体公开了该范围的上限和下限以及它们之间的每个中间值。在任何陈述值或陈述范围内的中间值以及任何其他陈述值或在所述范围内的中间值之间的每个较小的范围也包括在本发明内。这些较小范围的上限和下限可独立地包括或排除在范围内。
[0026]
除非另有说明,否则本文使用的所有技术和科学术语具有本发明所述领域的常规技术人员通常理解的相同含义。虽然本发明仅描述了优选的方法和材料,但是在本发明的实施或测试中也可以使用与本文所述相似或等同的任何方法和材料。本说明书中提到的所有文献通过引用并入,用以公开和描述与所述文献相关的方法和/或材料。在与任何并入的文献冲突时,以本说明书的内容为准。除非另有说明,否则“%”为基于重量的百分数。
[0027]
本发明提供用于检测基因融合的靶向测序方法,本文有时简称“本发明的靶向测
序方法”。其中,基因融合包括基因a和基因b两个基因之间的融合,还包括三个或更多个基因之间,例如三个基因之间的融合。本发明的基因融合优选不同基因的外显子之间发生的融合,不包括在来源于不同基因的外显子内部发生的融合。融合基因之间可相互称作对方的伴侣基因。
[0028]
本发明的靶向测序方法一般包括以下步骤:
[0029]
(1)预文库构建,其包括提取样本的总rna,反转录得到cdna,然后片段化为150-250bp的片段、并在末端修复、加a,连接测序接头,得到预文库;
[0030]
(2)利用封闭阻断剂封闭所述预文库,然后使探针组与所述预文库杂交,获得捕获片段,经pcr富集和纯化后质检,得到测序文库;
[0031]
(3)利用二代测序对所述测序文库进行测序,获得基因融合信息。
[0032]
本发明的步骤(1)为预文库构建步骤。其一般总rna提取、反转录为cdna、片段化和测序接头连接。可选地,在连接测序接头后进一步包括富集步骤。本发明设计的方法不会受到核糖体rna的影响。因此,在总rna进行文库构建时,可不必除去核糖体rna。本发明的片段化可用已知方法进行,例如离子片段化或超声波片段化,只要能够得到150-250bp的片段即可。
[0033]
本发明的步骤(2)为构建测序文库的步骤,其包括利用封闭阻断剂封闭预文库,然后使探针组与预文库杂交,从而获得捕获片段,随后经pcr富集和纯化后质检,得到测序文库。
[0034]
本发明的封闭阻断剂可使用已知任意阻断剂。优选使用发明人设计的通用阻断剂。本发明的通用阻断剂由两条含有天然核苷酸和人工核苷酸的寡核苷酸组成,并且不含有index序列(索引序列)。优选地,在通用阻断剂的3’端设计间隔臂,用于阻止3’端外切酶和3’端聚合酶发挥作用。优选地,人工核苷酸为锁核酸。锁核酸具有更高的tm值,通过控制锁核酸在通用阻断剂中的比例,可以方便地控制其形成的双链结构的稳定性。
[0035]
本发明中,探针组由能够与基因a各外显子的5’末端和3’末端互补的第一探针或能够与基因b各外显子的5’末端和3’末端互补的第二探针组成。需要说明的是,本文中“第一探针”和“第二探针”仅用于区分与基因a还是与基因b互补,并非用于表示探针种类。实际上,第一探针或第二探针分别包含多种不同类型探针的组合的含义。基因a和基因b并不一定表示编码产生完整全长蛋白的基因。实际上,基因a和基因b还可以分别表示编码完整蛋白某一部分的基因。此外,基因a和基因b中可以分别包含一个外显子。优选地,基因a和基因b中分别包含多个外显子,例如,分别包含2个、3个、5个,甚至更多个外显子。
[0036]
本发明中,对于基因a和基因b不特别限定,可以是任意可能发生融合的基因。基因a和基因b的实例包括但不限于abl1、akt1、akt3、alk、arhgap26、axl、braf、brd3、brd4、calca、camta1、ccnb3、ccnd1、cic、ctnnb1、ddr2、egfr、epc1、erbb2、erbb4、erg、esr1、esrra、etv1、etv4、etv5、etv6、ewsr1、fgfr1、fgfr2、fgfr3、fgr、foxo1、fus、gli1、gnas、hmga2、hras、idh1、idh2、insr、jak2、jazf1、kras、krt20、maml2、map2k1、mast1、mast2、meaf6、met、mkl2、msmb、musk、myb、ncoa2、notch1、notch2、nras、nrg1、ntrk1、ntrk2、ntrk3、numbl、pdgfb、pdgfra、pdgfrb、pik3ca、pkn1、plag1、pparg、prkca、prkcb、pth、ptk2b、raf1、rara、rela、ret、ros1、rspo2、rspo3、slc5a5、ss18、stat6、taf15、tcf12、tert、tfe3、tfeb、tfg、thada、tmprss2、ttf1和ywhae种基因。优选地,基因a和基因b选自alk、ret、ros1、
ntrk1、ntrk2、ntrk3、braf、pik3ca、met、fgfr1、fgfr2、fgfr3、prkca、prkcb、kras、jak2、akt1和akt3。
[0037]
具体而言,第一探针包含一或多条探针,每条探针各自分别能够与基因a中某一外显子的5’末端或3’末端互补,第一探针中的多条探针使得基因a中各外显子的5’末端和3’末端分别能够与对应的一条探针互补结合。类似地,第二探针包含一或多条探针,每条探针各自分别能够与基因b中某一外显子的5’末端或3’末端互补,第二探针中的多条探针使得基因b中各外显子的5’末端和3’末端分别能够与对应的一条探针互补结合。
[0038]
在示例性实施方案中,在基因a包含第一外显子和第二外显子,基因b包含第三外显子和第四外显子的情况下,融合事件可能发生于第一外显子3’端和第三外显子5’端之间、第一外显子3’端和第四外显子5’端之间、第一外显子5’端和第三外显子3’端之间、第一外显子5’端和第四外显子3’端之间、第二外显子3’端和第三外显子5’端之间、第二外显子3’端和第四外显子5’端之间、第二外显子5’端和第三外显子3’端之间、第二外显子5’端和第四外显子3’端之间。
[0039]
本发明中,外显子的5’末端是指其5’端第1个碱基至第120个碱基之间的片段,优选第1个碱基至第110个碱基之间的片段,更优选第1个碱基至第100个碱基之间的片段,还优选第1个碱基至第90个碱基之间的片段。外显子的3’末端是指其3’端最后1个碱基至倒数第120个碱基之间的片段,优选3’端最后1个碱基至倒数第110个碱基之间的片段,更优选3’端最后1个碱基至倒数第100碱基之间的片段,还优选3’端最后1个碱基至倒数第90个碱基之间的片段。本发明中,同一外显子的5’末端和3’末端的长度之和一般小于整个外显子的长度。优选5’末端和3’末端的长度之和小于外显子整体长度的一半,更优选小于外显子整体长度的2/3,甚至3/4。本发明的探针组中,各探针仅针对外显子的5’末端和3’末端,因此大大降低了探针的数量。
[0040]
为了进一步提高探针与目标序列的结合力,本发明进一步优化捕获探针的标记物。优选地,各探针(包括第一探针和第二探针中的各探针)分别为5’端生物素修饰的探针。更优选地,生物素分子通过间隔臂与探针5’端的羟基连接。优选地,间隔臂具有下式(i)的结构-nh(ch2)mo-(-c=o-nh-(ch
2-ch2o-)4)n,其中m为4-8的整数,优选为5-6的整数,n为1-5的整数,优选2-3的整数。本发明发现通过使用式(i)作为间隔臂,可以大大提高靶向测序时的捕获效率。
[0041]
在本发明中,对于100-120nt探针,优选60-70℃杂交及清洗;对于80-100nt探针,优选55-65℃杂交及清洗;对于60-80nt探针,优选50-60℃进行杂交及清洗。
[0042]
本发明中,在步骤(2)的杂交步骤中,预文库可以是一个,也可以为多个不同的预文库的混合文库。
[0043]
本发明的步骤(3)为利用二代测序技术对测序文库进行测序,获得基因融合信息的步骤。二代测序技术有时也称作高通量测序技术,是一次并行对几十万到几百万条dna分子进行序列测定,又称下一代测序技术。二代测序技术的核心思想是边合成边测序,即通过捕捉新合成的末端的标记来确定dna的序列。二代测序技术的实例包括illumina测序技术和life tech(thermo scientific)测序技术。
[0044]
实施例1
[0045]
本实施例研究探针与目的片段的结合长度对探针结合力的影响。
[0046]
一、探针选择:
[0047]
为了检测探针与目的片段的结合长度对探针结合力的影响,采用尺寸约为100kb的人类基因组探针池进行捕获测序,随后选择上下游1kb内无其他探针,gc含量为47.5-52.5%的16条探针对应区间进行分析。
[0048]
表1
[0049]
[0050][0051]
二、预文库构建及捕获测序:
[0052]
采用dna建库试剂盒(rapid dna lib prep kit,abclonal)对na12878 gdna(coriell)构建本实施例所用的预文库(插入片段大小:~200bp;pcr循环数:7)。
[0053]
按照步骤如a-j所示进行4小时杂交捕获。
[0054]
a.文库预封闭
[0055]
将表2试剂加入到0.2ml低吸附离心管(eppendorf)中,使用真空浓缩仪(eppendorf)将离心管中溶液蒸干备用。
[0056]
表2
[0057][0058]
b.探针与文库杂交
[0059]
将13μl杂交缓冲液(0.33m sodium phosphate buffer ph7.0、0.65%sds(w/v)、1.31mm edta、1.31x ssc、2.62x denhardt’s solution、20%甲酰胺(v/v))加入到上述步骤的离心管中,涡旋混匀,室温孵育5分钟。
[0060]
95℃变性10分钟,随后加入4μl(3pmol)探针池,涡旋混匀,65℃孵育4小时。
[0061]
c.清洗液准备
[0062]
按照表3所示准备清洗缓冲液,其中,1x wash buffer s和部分1x wash buffer i在65℃条件下预热30分钟后使用。
[0063]
表3
[0064][0065][0066]
1x beads wash buffer:1m nacl、10mm tris-hcl ph 7.5、1mm edta、0.1%(v/v)tween-20
[0067]
1x wash buffer s:1x ssc、0.1%(v/v)tween-20,ph7.0
[0068]
1x wash buffer i:1x ssc、0.1%(w/v)sds,ph7.0
[0069]
1x wash buffer ii:0.5x ssc,ph7.0
[0070]
1x wash buffer iii:0.2x ssc,ph7.0
[0071]
d.链霉亲和素磁珠准备
[0072]
将链霉亲和素磁珠(dyna beads m270,invitrogen)从冰箱中(4℃)取出恢复到室温(约30分钟)。涡旋混匀15秒。取100μl链霉亲和素磁珠加入到新的1.5ml低吸附离心管中。将离心管放到磁力架上,直到溶液澄清。吸弃上清,切勿扰动磁珠。按以下步骤对链霉亲和素磁珠进行清洗:
[0073]
(1)将离心管从磁力架上取下,加入200μl 1x beads wash buffer,涡旋振荡10秒。
[0074]
(2)将离心管瞬时离心,放到磁力架上,直到溶液澄清,吸弃上清,切勿扰动磁珠。
[0075]
重复步骤(1)和(2)。
[0076]
将离心管从磁力架上取下,加入100μl 1x beads wash buffer。将离心管中的100μl磁珠重悬液转移到新的0.2ml低吸附离心管(eppendorf)中待用。将离心管放到磁力架上,直到溶液澄清。吸弃上清,切勿扰动磁珠,立即进行后续实验步骤。
[0077]
e.链霉亲和素磁珠捕获
[0078]
将杂交混合物加入到含链霉亲和素磁珠的0.2ml低吸附离心管中。使用移液器轻柔吹吸10次混匀。使用pcr仪(热盖温度设置为75℃)65℃孵育45分钟。每12分钟涡旋混匀3秒,确保磁珠处于悬浮状态。
[0079]
f.捕获后清洗
[0080]
1.65℃清洗步骤:
[0081]
将100μl预热的1x wash buffer i加入到含有杂交混合物的0.2ml低吸附离心管中。吹吸混匀后,将含有链霉亲和素磁珠的反应液转移到新的1.5ml低吸附离心管中。将离心管放置到磁力架上,直到溶液澄清,吸弃上清。
[0082]
继续按以下步骤进行清洗:
[0083]
(1)加入200μl预热的1x wash buffer s,吹吸或涡旋混匀后,在65℃条件下孵育5分钟。
[0084]
(2)瞬时离心,将离心管放置到磁力架上,直到溶液澄清,吸弃上清。
[0085]
重复步骤(1)和(2)。
[0086]
2.室温清洗
[0087]
加入200μl1x wash buffer i,涡旋混匀2分钟。将离心管瞬时离心,放置到磁力架上,直到溶液澄清,吸弃上清。加入200μl1x wash buffer ii,涡旋混匀1分钟。将离心管瞬时离心,放置到磁力架上,直到溶液澄清,吸弃上清。加入200μl1x wash buffer iii,涡旋混匀30秒。将离心管瞬时离心,放置到磁力架上,直到溶液澄清,吸弃上清。
[0088]
3.磁珠重悬
[0089]
立即加入20μl无酶无菌水。使用移液器吹吸10次,重悬磁珠,进入后续实验步骤。
[0090]
g.pcr扩增
[0091]
按照表4配置pcr反应体系。
[0092]
表4
[0093][0094]
吹吸或低速涡旋混匀使磁珠保持悬浮状态,立即进入pcr步骤。使用pcr仪按表5程序运行,热盖温度105℃。
[0095]
表5
[0096][0097]
h.pcr产物纯化
[0098]
每个pcr管中加入75μl ampure xp纯化磁珠(beckman)。按照ampure xp操作手册纯化pcr产物。使用22μl tris-hcl(10mm,ph 8.5)进行洗脱。转移20μl包含捕获文库的洗脱液到新的1.5ml低吸附离心管(eppendorf)中。
[0099]
i.文库质控
[0100]
使用qubit荧光计3.0(thermofisher)测量文库浓度。使用agilent 2100测量文库片段长度,产物集中在320bp之间,无接头二聚。
[0101]
j.高通量测序
[0102]
采用illumina novaseq测序仪进行pe150模式测序。
[0103]
三、数据分析
[0104]
使用trimmomatic去除接头以及低质量序列得到clean data,然后使用samtools提取目标区域的reads,根据比对位置,分析探针与dna结合大小。
[0105]
cap01和cap02的测序深度分别为12948x和13271x,使用110-119bp结合长度所捕获的目的片段数目平均值作为对照,分别统计1-120bp结合长度所捕获目的片段与上述对照的比值。
[0106]
如图3所示,探针捕获片段的最短结合长度约为40bp,其结合能力随着结合长度逐渐增加,在70bp左右达到50%,并在100bp左右达到饱和,进入平台期。因此,对于120nt探针,保证探针与目的片段的结合长度超过100bp,将更有效的捕获目标序列。而对于短长度结合(<70bp),探针对目的片段的捕获将会产生较大影响。
[0107]
实施例2
[0108]
本实施例为融合基因检测例。
[0109]
一、探针设计及合成:
[0110]
分别采用传统方法和本发明方法对90个肿瘤基因融合突变相关的基因设计探针,部分基因信息如下表6所示;以alk为例,图4示出了传统方法和本发明方法对alk进行探针
设计的区别,二者分别使用39和29条120nt探针,覆盖4614bp和3235bp的区间;对于上述90个基因,传统方法和本发明方法分别使用2075条和1405条120nt探针;其中,alk、ret和ros1基因的具体转录本和探针信息如下所示。
[0111]
表6传统探针设计
[0112]
[0113]
[0114]
[0115]
[0116][0117][0118]
表7本发明的探针设计
[0119]
[0120]
[0121]
[0122][0123]
除此之外,如图5所示,对已知融合亚型设计融合探针,用于进一步提高融合检测灵敏度,alk、ret和ros1涉及的融合探针序列如下表8所示。
[0124]
表8
[0125]
[0126]
[0127][0128]
二、预文库构建:
[0129]
采用rna建库试剂盒(mrna-seq lib prep kit for illumina,abclonal)对含有alk、ret和ros1融合基因的5份总rna构建预文库(rna投入量:100ng;插入片段大小:~200bp;pcr循环数:10)。
[0130]
分别采用传统设计探针、本发明设计探针以及加入融合探针的本发明设计探针,分别对上述5份预文库进行杂交捕获,然后进行二代测序、信息分析。捕获测序流程同实施例1。
[0131]
三、数据分析
[0132]
使用trimmomatic去除接头以及低质量序列得到clean data,然后使用star进行序列比对,最后使用star-fusion进行融合基因分析。如表9所示,数据分析表明,采用本发明设计的探针,其中靶率与传统方法相近,无明显差异;同时,由于使用更少的探针,覆盖更少的区间,因此,本发明使用更少的数据量,便能够获得与传统设计接近的测序深度。
[0133]
表9
[0134]
[0135][0136]
进一步的融合突变分析表明,本发明方法所检测到的alk、ret和ros1融合基因的阳性reads数是传统设计的1.4-2.1倍,灵敏度更高;而在加入融合探针后,阳性reads数目进一步提升到本发明设计的1.5-3.6倍,灵敏度进一步提升(表10)。上述结果表明,本发明的探针设计方案,使用更少的探针和更低的测序数据量,能够获得更高的检测灵敏度,而加入融合探针,可以更高效的检测已知的融合基因亚型。
[0137]
表10
[0138][0139][0140]
综上所述,本发明具有通量高、灵敏度高、捕获效率高、成本低的优势。与qpcr等传统技术一次只能检测一种融合亚型相比,本发明能够一次检测多个基因的“宽度”,将大大缩减诊断时间。此外,本发明还能够发现新的融合亚型,不需要融合基因的先验知识,同时,
加入融合探针,可以进一步提升已知融合亚型的检测灵敏度。可以预见,随着针对融合基因的肿瘤靶向药物的逐渐丰富,融合基因的数量也将增多,qpcr等传统技术将不能满足融合基因的检测需要,本发明在融合基因的快速、准确诊断中,具有极高的应用价值和潜能。
[0141]
尽管本发明已经参考示例性实施方案进行了描述,但应理解本发明不限于公开的示例性实施方案。在不背离本发明的范围或精神的情况下,可对本发明说明书的示例性实施方案做多种调整或变化。权利要求的范围应基于最宽的解释以涵盖所有修改和等同结构与功能。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1