一种无溶剂配位稀土MOF材料及其制备方法和应用
一种无溶剂配位稀土mof材料及其制备方法和应用
技术领域
1.本发明属于医药技术领域,具体涉及一种无溶剂配位稀土mof材料及其制备方法和应用,尤其涉及一种无溶剂配位的可见/红外发光的mof材料及其制备方法和应用。
背景技术:
2.近红外(nir)发光材料已在光通信、生物成像、传感、激光以及太阳能转换等各种应用中显示出巨大应用前景。近红外发光通常是通过使用镧系离子(ln
3+
)获得的;其中,yb
3+
、nd
3+
和er
3+
是显示近红外发射的最常用的镧系元素。ln
3+
的窄带宽的发射,对光漂白的高抗性,以及发射带的位置对环境条件的独立性等固有特点使其在实际应用中具有很大的吸引力。
3.近年来,近红外发射的金属有机框架(mofs)因其独特的性能而备受关注,它将mof平台的高孔隙率和结构可调性与镧系元素的发光结合起来,形成了新型的多功能材料。到目前为止,已经有几十种基于yb
3+
、nd
3+
和er
3+
的mofs报告了近红外发光,还有一些使用不太常见的近红外发光的ln
3+
,如pr
3+
、ho
3+
和sm
3+
。镧系mofs通过"天线效应"表现出发光,在光的照射下,发色配体吸收大量的光子能量,并将相应的能量转移到镧系离子的接受电子层。这种效应可以解决镧系离子的低摩尔消光系数(《10m-1
cm-1
)的限制,当镧系化合物被直接激发时,往往导致非常低的发光强度。值得注意的是,由于非辐射弛豫作用,当镧系离子靠近配体和配位溶剂上的n-h、o-h和c-h基团时,近红外发光发射会被明显抑制。为了进一步增加近红外发射,通过使用全氟和过氘配体将c-h键替换为能量较低、淬灭较少的基团,如c-f和c-d,是一种众所周知的方法。
4.在mof领域,设计高量子产率的近红外发射mof的努力相当罕见。在非专利文献1(《inorganic chemistry》,2006年,第45卷,第8882-8886页)报道了通过将配体1,4-苯二甲酸酯(bdc
2-)替换为其氟化对应物四氟对苯二甲酸酯(f
4-bdc
2-),可以实现er
3+
基mof的近红外发射(λex=808nm)的增强。
5.在非专利文献2(《journal of materials chemistry a》,2020年,第8卷,第10188
–
10192页)报道了利用使用配体空间位阻,能够获得无溶剂配位的、高发射率的新型re-mof。
6.因此,设计高近红外发射mofs的更直接的方法应该首先解决溶剂协调问题。这代表了一种合成挑战,因为re
3+
离子的尺寸很大,导致小溶剂分子结合的概率很高。事实上,在大多数报道的稀土mof中,re
3+
的配位中存在配位的溶剂分子是很常见的。
7.此前,各种方法,包括微波辅助合成、水/溶剂热合成和界面自组装策略,已经被开发出来,以优化合成系统,在不同的纳米或微尺度上定制mofs的结晶度和生长。然而,这些策略通常会带来严重的溶剂配位问题。
8.最近,机械化学研磨合成作为一种无溶剂的绿色合成策略,为快速和大规模合成发光的re-mof材料开辟了一条新途径。是解决溶剂配位问题的潜在方法之一。
9.综上,由于re
3+
离子的大尺寸,导致溶剂小分子配位极其普遍。设计高nir发射mof
的更直接方法应该首先解决溶剂配位问题。
10.因此,开发一种无溶剂的绿色合成策略来解决溶剂配位问题,对制备高红外发射稀土mof材料是非常重要的。
技术实现要素:
11.本发明目的在于提供一种无溶剂配位稀土mof材料及其制备方法和应用,尤其涉及一种无溶剂配位的可见/红外发光的mof材料及其制备方法和应用;利用无溶剂法机械化学法和热处理方法,制备出新型无溶剂配位的稀土mof材料,该无溶剂配位稀土mof材料在可见/红外波段发光。本发明所涉及新型无溶剂配位稀土mof材料的制备,方法简便,产率高,条件相对温和,具有大规模生产的前景;其中多组分无溶剂配位稀土mof材料具有优异的可见、红外荧光性能,有望在光、电传感、生物成像领域得到应用。
12.本发明的目的之一在于提供一种无溶剂配位稀土mof材料,所述无溶剂配位稀土mof材料的分子式为re(c9h3o6),其中re为sc、y、la、ce、pr、nd、sm、eu、gd、tb、dy、ho、er、tm、yb或lu中的任意一种或至少两种的组合;
13.所述无溶剂配位稀土mof材料具有如下式(i)结构:
[0014][0015]
式(i)中,re相同或不同。
[0016]
优选地,所述无溶剂配位稀土mof材料的结构晶体学特征包括:空间群为r3c,晶胞参数为α=90
°
,β=90
°
,γ=120
°
;
[0017]
当re以yb为例,所述无溶剂配位稀土mof材料的结构晶体学特征包括:空间群为r3c,晶胞参数为α=90
°
,β=90
°
,γ=120
°
。以sc为例,所述无溶剂配位稀土mof材料的结构晶体学特征包括:空间群为r3c,晶胞参数为剂配位稀土mof材料的结构晶体学特征包括:空间群为r3c,晶胞参数为α=90
°
,β=90
°
,γ=120
°
。
[0018]
优选地,所述无溶剂配位稀土mof材料的x-粉末衍射谱图中,在2θ衍射角为14.8
±
0.2
°
、19.8
±
0.2
°
、22.3
±
0.2
°
、24.9
±
0.2
°
、30.0
±
0.2
°
、32.2
±
0.2
°
、35.2
±
0.2
°
、36.2
±
0.2
°
处有特征峰。或在2θ衍射角为15.3
±
0.2
°
、20.6
±
0.2
°
、22.6
±
0.2
°
、25.2
±
0.2
°
、25.7
±
0.2
°
、29.0
±
0.2
°
、30.8
±
0.2
°
、32.0
±
0.2
°
处有特征峰。
[0019]
优选地,所述无溶剂配位稀土mof材料的红外光谱在2500-2500区间无配位溶剂的特征峰。
[0020]
优选地,所述无溶剂配位稀土mof材料的荧光发射光在300nm激发下,有可见、红外荧光特性。
[0021]
本发明的目的之二在于提供一种如目的之一所述的无溶剂配位稀土mof材料的制备方法,所述制备方法包括如下步骤:
[0022]
将re(no3)3.nh2o和均苯三甲酸混合均匀,而后热处理得到所述无溶剂配位稀土mof材料。
[0023]
优选地,所述re为sc、y、la、ce、pr、nd、sm、eu、gd、tb、dy、ho、er、tm、yb或lu中的任意一种或至少两种的组合;
[0024]
优选地,所述re(no3)3.nh2o和均苯三甲酸的摩尔比为1:1;
[0025]
优选地,所述混合的方式为研磨;
[0026]
优选地,所述混合的时间为2-10min。
[0027]
优选地,所述热处理的温度为120-160℃,热处理的时间为12-48h;
[0028]
优选地,所述热处理是在聚四氟乙烯中进行的。
[0029]
优选地,所述制备方法还包括将热处理后得到的混合物依次进行清洗和干燥;
[0030]
优选地,所述清洗包括先用蒸馏水洗涤3-5次,再用乙醇洗涤3-5次;
[0031]
优选地,所述干燥为真空干燥,所述真空干燥的真空度为0.05~0.1mpa,干燥的温度为50~80℃,干燥的时间为2~12h。
[0032]
本发明的目的之三在于根据目的之一所述无溶剂配位稀土mof材料在可见/红外光致发光领域的应用。
[0033]
本发明的技术特点和有益效果:
[0034]
本发明利用无溶剂法机械化学法和热处理方法,制备出新型无溶剂配位的稀土mof材料,该无溶剂配位稀土mof材料在可见/红外波段发光。本发明所涉及新型无溶剂配位稀土mof材料的制备,方法简便,产率高,条件相对温和,具有大规模生产的前景;其中多组分无溶剂配位稀土mof材料具有优异的可见、红外荧光性能,有望在光、电传感、生物成像领域得到应用。
附图说明
[0035]
图1为本发明涉及的由具体实施例15所合成re-mofs样品的结构图。
[0036]
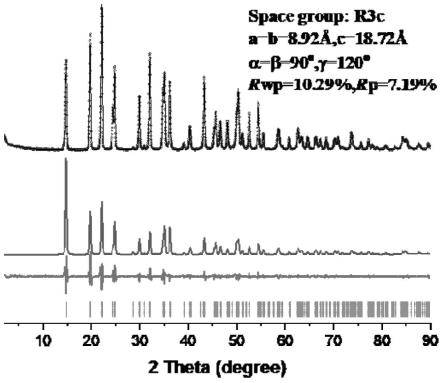
图2为本发明涉及的由具体实施例1所制备的分级结构块状eu-mofs的p-xrd精修图。
[0037]
图3为本发明涉及的由具体实施例1、2、3、4、5、6、7、8、9、10、11、12、13、14、15和16所制备样品的xrd图。
[0038]
图4为本发明涉及的由具体实施例1、2、3、4、5、6、7、8、9、10、11、12、13、14、15和16所制备样品的红外光谱图。
[0039]
图5为本发明涉及的由具体实施例17、18、19、20、21、22和23所合成多组分re-mofs在300nm激发波长下的荧光可见、红外发射谱图。
具体实施方式
[0040]
实施例1:
[0041]
0.5-5mmol的sc(no3)3.6h2o和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得sc-mofs材料,样品命名为样品1。
[0042]
实施例2:
[0043]
0.5-5mmol的y(no3)3.6h2o和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得y-mofs材料,样品命名为样品2。
[0044]
实施例3:
[0045]
0.5-5mmol的la(no3)3.6h2o和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得la-mofs材料,样品命名为样品3。
[0046]
实施例4:
[0047]
0.5-5mmol的ce(no3)4.6h2o和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得ce-mofs材料,样品命名为样品4。
[0048]
实施例5:
[0049]
0.5-5mmol的pr(no3)3.6h2o和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得pr-mofs材料,样品命名为样品5。
[0050]
实施例6:
[0051]
0.5-5mmol的nd(no3)3.6h2o和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得nd-mofs材料,样品命名为样品6。
[0052]
实施例7:
[0053]
0.5-5mmol的sm(no3)3.6h2o和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得sm-mofs材料,样品命名为样品7。
[0054]
实施例8:
[0055]
0.5-5mmol的eu(no3)3.6h2o和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得eu-mofs材料,样品命名为样品8。
[0056]
实施例9:
[0057]
0.5-5mmol的gd(no3)3.6h2o和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得gd-mofs材料,样品命名为样品9。
[0058]
实施例10:
[0059]
0.5-5mmol的tb(no3)3.6h2o和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得tb-mofs材料,样品命名为样品10。
[0060]
实施例11:
[0061]
0.5-5mmol的dy(no3)3.6h2o和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得dy-mofs材料,样品命名为样品11。
[0062]
实施例12:
[0063]
0.5-5mmol的ho(no3)3.6h2o和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得ho-mofs材料,样品命名为样品12。
[0064]
实施例13:
[0065]
0.5-5mmol的er(no3)3.6h2o和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得er-mofs材料,样品命名为样品13。
[0066]
实施例14:
[0067]
0.5-5mmol的tm(no3)3.6h2o和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得tm-mofs材料,样品命名为样品14。
[0068]
实施例15:
[0069]
0.5-5mmol的yb(no3)3.6h2o和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得yb-mofs材料,样品命名为样品15。
[0070]
实施例16:
[0071]
0.5-5mmol的lu(no3)3.6h2o和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在
60℃下真空干燥2小时,即可获得lu-mofs材料,样品命名为样品16。
[0072]
实施例17:
[0073]
0.25-2.5mmol的sc(no3)3.6h2o、0.25-2.5mmol的dy(no3)3.6h2o(金属掺杂比1:1)和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得dy/sc-mofs材料,样品命名为样品17。
[0074]
实施例18:
[0075]
0.25-2.5mmol的sc(no3)3.6h2o、0.25-2.5mmol的tb(no3)3.6h2o(金属掺杂比1:1)和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得tb/sc-mofs材料,样品命名为样品18。
[0076]
实施例19:
[0077]
0.25-2.5mmol的sc(no3)3.6h2o、0.25-2.5mmol的eu(no3)3.6h2o(金属掺杂比1:1)和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得eu/sc-mofs材料,样品命名为样品19。
[0078]
实施例20:
[0079]
0.25-2.5mmol的sc(no3)3.6h2o、0.25-2.5mmol的sm(no3)3.6h2o(金属掺杂比1:1)和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得sm/sc-mofs材料,样品命名为样品20。
[0080]
实施例21:
[0081]
0.25-2.5mmol的sc(no3)3.6h2o、0.25-2.5mmol的yb(no3)3.6h2o(金属掺杂比1:1)和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得yb/sc-mofs材料,样品命名为样品21。
[0082]
实施例22:
[0083]
0.25-2.5mmol的sc(no3)3.6h2o、0.25-2.5mmol的nd(no3)3.6h2o(金属掺杂比1:1)和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得nd/sc-mofs材料,样品命名为样品22。
[0084]
实施例23:
[0085]
0.25-2.5mmol的sc(no3)3.6h2o、0.25-2.5mmol的er(no3)3.6h2o(金属掺杂比1:1)
和0.5-5mmol的h3btc(均苯三甲酸)在研钵研磨2-10分钟,至均匀。然后将混合物转移至25ml高压釜中,并在120℃-160℃加热12-48小时,至不溶水的白色固体生成。冷却至室温后,收集产物并用蒸馏水洗涤3次,再用乙醇洗涤3次,并在60℃下真空干燥2小时,即可获得er/sc-mofs材料,样品命名为样品23。
[0086]
本发明对制备的样品进行一系列(xrd、红外光谱、荧光光谱)表征分析以证实我们设计的方法成功合成了新型无溶剂配位的稀土mof样品,具有良好的可见/红外发光性质。
[0087]
从图1的结构图可知,该mof为每个金属原子有6个配体配位,每个配体配位6个原子。
[0088]
从图2的p-xrd精修图可知,利用p-xrd在materials studio中搭建结构,拟合程度达rp=7.19%。
[0089]
从图3的xrd图可知,由实施例1、2、3、4、5、6、7、8、9、10、11、12、13、14、15和16所制备的样品为re-mofs,其中1、15、16为新型稀土mof。
[0090]
从图4的红外光谱图可知,新型re-mofs的红外显示没有水分子的伸缩振动吸收峰,证明该新型mof无溶剂配位。
[0091]
从图5荧光发射光谱看出,在300nm激发下,样品17、18、19、20、21、22和23有优异的可见、红外荧光特性。
[0092]
本发明的方法与产品已经通过较佳实施例子进行了相关描述,相关技术人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法和产品进行改动或适当的变更与组合,来实现本发明技术。特别需要指出的是,所有相似的替换和改动对本领域技术人员来说是显而易见的,他们都被视为包括在本发明的精神、范围和内容中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1