具有肿瘤靶向的生物素化金(I)配合物及其制备方法和应用
具有肿瘤靶向的生物素化金(i)配合物及其制备方法和应用
技术领域
1.本发明属于药物制备领域,具体涉及具有肿瘤靶向的生物素化金(i)配合物及其制备方法和应用。
背景技术:
2.放射治疗是利用放射线治疗肿瘤的一种治疗方法,放射线包括放射性同位素产生的α、β、γ射线和各类x射线治疗机或加速器产生的x射线、电子线、质子束及其他粒子束等。放射治疗是仅次于手术的恶性肿瘤第二大治疗手段,但临床上常因正常组织耐受剂量的限制而不能给予肿瘤足够的照射剂量,最终导致治疗失败,因此,如何提高肿瘤对射线的敏感性是临床肿瘤放疗面临的突出问题。
3.放射增敏剂作为一种增强肿瘤放疗敏感性、提高放疗疗效的药物,通过增加辐射诱导的氧自由基及dna损伤、调控放疗关键分子靶点以达到放射增敏目的。尽管放疗增敏剂经过了数十年的发展,已开发出许多种类,但相关研究仍无法满足放疗的临床需要。例如,顺铂是目前普遍用于治疗宫颈癌的铂增敏剂之一,尽管以顺铂为基础的同步化疗和放疗已被证明能提高宫颈癌患者的总体生存率和无进展生存率,降低复发率,改善预后,增加手术机会,减少术中和术后并发症。但众所周知,铂基金属药物具有许多缺点,如毒性高、选择性低和耐药性。更为关键的是,现有的许多放射增敏剂的临床应用并未完全得到肯定,尚需要明确其作用靶点和机制,进一步优化放射增敏剂,从而成为更有效的临床辅助手段。因此,开发靶点和作用机制明确的放疗增敏剂是下一代放疗增敏剂研发的重要方向之一。
4.综上,开发一种有效增强放疗敏感性且靶点和作用机制明确的放射增敏剂是本领域亟待解决的问题。
技术实现要素:
5.本发明为解决传统放射增敏剂选择性低、机制不明确等缺点,公开了一种具有肿瘤靶向的生物素化金(i)配合物及其制备方法和应用,该生物素化金(i)配合物可实现靶向肿瘤细胞、抑制肿瘤细胞增殖和增强放疗敏感性的多重效能,且选择性高,具有较好的应用前景。
6.为解决上述问题,本发明采用以下技术方案实现:
7.本发明首先提供了一种具有肿瘤靶向的生物素化金(i)配合物,其结构如式i所示:
8.9.其中,x为
10.r为pta或pph3,结构如下所示:
[0011][0012]
本发明另一方面还提供了一种前面所述的具有肿瘤靶向的生物素化金(i)配合物的制备方法,包括以下步骤:
[0013]
(1)中间体的制备:
[0014]
中间体2a:以生物素和4-乙炔基苯胺为原料,加入缩合剂,避光,氮气保护下室温搅拌反应,反应液经过稀释、萃取、洗涤有机层、干燥、过滤后,进一步进行浓缩和分离纯化,得到中间体2a,结构如下所示:
[0015][0016]
中间体2b:以生物素和炔丙基胺为原料,加入缩合剂,室温条件下搅拌反应,反应液经过浓缩、复溶、过滤后,进一步进行浓缩和分离纯化,得到中间体2b,结构如下所示:
[0017][0018]
(2)生物素化金(i)配合物3a-3d的制备:
[0019]
配合物3a:以ph3paucl、所述中间体2a和碱为原料,避光、氮气保护下室温搅拌反应,反应结束后,反应液经过滤、洗涤,得到化合物3a;
[0020]
配合物3b:以aupta、所述中间体2a和碱为原料,避光、氮气保护下室温搅拌反应,反应结束后,反应液经过滤、洗涤,得到化合物3b;
[0021]
配合物3c:以aupta、所述中间体2b和碱为原料,避光、氮气保护下室温搅拌反应,反应结束后,反应液经过滤、洗涤、纯化分离,得到化合物3c;
[0022]
配合物3d:以ph3paucl、所述中间体2b和碱为原料,避光、氮气保护下室温搅拌反应,tlc检测反应完全后,加水终止反应,反应液经萃取、干燥、过滤、蒸发、纯化分离后,得到化合物3d;
[0023]
其中,配合物3a-3d的结构式如下所示:
[0024][0025]
优选地,所述的碱包括碳酸钾、氢氧化钠、氢氧化钾或碳酸钠中的任意一种。
[0026]
本发明另一方面还提供了一种前面所述的具有肿瘤靶向的生物素化金(i)配合物或其可药用的盐在制备抗肿瘤药物中的应用。
[0027]
优选地,所述的肿瘤包括宫颈癌。
[0028]
优选地,所述的生物素化金(i)配合物或其可药用的盐用于抑制肿瘤细胞trxr活性,从而产生ros,导致肿瘤细胞线粒体功能受损、细胞周期停滞和细胞凋亡。
[0029]
本发明另一方面还提供了一种前面所述的具有肿瘤靶向的生物素化金(i)配合物或其可药用的盐在制备放射增敏剂中的应用。
[0030]
优选地,所述的生物素化金(i)配合物或其可药用的盐通过靶向结合并抑制trxr干预氧化还原稳态,并诱导铁死亡过程,从而使肿瘤细胞对放疗敏感。
[0031]
相对于现有技术,本发明的有益效果是:
[0032]
1、本发明公开的生物素化金(i)配合物对肿瘤细胞具有靶向特异性,且能靶向结合并抑制trxr,产生大量的ros,导致肿瘤细胞线粒体功能受损、细胞周期停滞和细胞凋亡,具有抑制肿瘤细胞增殖的能力,更重要的是,肿瘤细胞处于g2/m期时,对放疗的敏感性增强;细胞内ros水平的升高,也可提高肿瘤细胞的固有辐射敏感性,增强放射线对肿瘤细胞的敏感性,实现肿瘤放疗增敏目的。
[0033]
2、本发明通过实验验证了所述的生物素化金(i)配合物3a具有高效放射增敏和选择性,其通过靶向trxr干预氧化还原稳态,并诱导铁死亡过程,实现增强辐射敏感的目的,为开发新型肿瘤放射治疗药物开辟新的途径。
附图说明
[0034]
图1为配合物3a的核磁共振谱图;
[0035]
图2为配合物3b的核磁共振谱图;
[0036]
图3为配合物3c的核磁共振谱图;
[0037]
图4为配合物3d的核磁共振谱图;
[0038]
图5为配合物3e的核磁共振谱图;
[0039]
图6为配合物3f的核磁共振谱图;
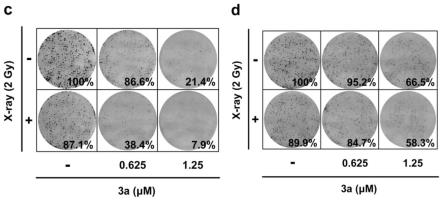
[0040]
图7为配合物3g的核磁共振谱图;
[0041]
图8为配合物3h的核磁共振谱图;
[0042]
图9为配合物3a-3h的hplc谱图;
[0043]
图10为hela、h8细胞对金配合物的细胞摄取对比图;
[0044]
图11为配合物3a对trxr抑制活性的结果;其中:
[0045]
a表示配合物3a对纯化的trxr酶的抑制活性;
[0046]
b表示采用配合物3a处理72小时后对hela细胞中trxr活性的影响;
[0047]
c表示免疫荧光检测hela细胞中配合物3a对trxr的抑制作用;
[0048]
d表示不同处理后hela细胞中trxr蛋白的表达水平;
[0049]
图12为为配合物3a与ct-dna的作用研究;其中:
[0050]
a-b表示配合物3a与ct-dna作用的紫外光谱图;
[0051]
c表示配合物3a与ct-dna作用的圆二色谱图;
[0052]
图13为配合物3a对细胞内ros水平和线粒体跨膜通透性变化的影响;其中:
[0053]
a表示荧光探针检测配合物3a对hela细胞中ros的影响;
[0054]
b表示配合物3a处理hela细胞后用jc-1染色的线粒体膜电位的荧光图;
[0055]
c表示流式细胞术测定不同处理方法对hela细胞中ros水平的影响;
[0056]
d表示流式细胞术测定不同处理方法对hela细胞中mmp水平的影响;
[0057]
图14为配合物3a处理对hela细胞凋亡和细胞周期的影响;其中:
[0058]
a表示配合物3a诱导hela细胞凋亡;
[0059]
b表示hela细胞中的蛋白质印迹分析;
[0060]
c表示配合物3a诱导hela细胞周期阻滞;
[0061]
图15为通过mtt检测的细胞存活率图(用nac预处理);
[0062]
图16为配合物3a的放射增敏实验结果,其中:
[0063]
a表示不同浓度3a和不同剂量x射线处理后的细胞活力对比;
[0064]
b表示x射线和3a处理对hela细胞的协同抗增殖作用的组合指数图分析;
[0065]
c-d表示3a和x射线处理15天后hela和h8细胞集落形成的代表性图像;
[0066]
图17表示配合物3a的放射增敏机制研究;其中:
[0067]
a表示不同浓度3a和x射线处理后的细胞活力对比;
[0068]
b表示不同处理后hela细胞中γ-h2ax蛋白的表达水平;
[0069]
c-d表示分别通过cck8测定法测定ferrostatin-1在0gy和2gy时的抵消作用;
[0070]
e表示不同处理后hela细胞中gpx-4蛋白的表达水平;
[0071]
图18表示ir和3a联合治疗对斑马鱼发育和生存的影响;其中:
[0072]
a-c表示斑马鱼发育过程中的血管形成和形态图;
[0073]
d-g表示不同处理后的胚胎孵化率和成活率;
[0074]
图19表示配合物3a的体内抗肿瘤研究结果,其中:第0天表示治疗前,第1~3天表
示治疗后。
具体实施方式
[0075]
以下结合附图和实施例对本发明的技术方案做进一步的说明。
[0076]
除非特别指明,本发明实施例中的试剂均可从正规渠道商购获得。
[0077]
除非特别说明,本发明实施例中的hela细胞、siha细胞和h8细胞购自中科院细胞库。
[0078]
本发明为解决传统放射增敏剂选择性低、机制不明确等缺点,设计了一系列全新的生物素化金(i)配合物,在设计时,基于以下几点考虑:
[0079]
(1)作为生物素的肿瘤靶向部分可连接到新型au(i)配合物,显著改善癌细胞的靶向性和摄取。
[0080]
(2)使用了不同类型的炔基配体和三重键双碳(-c≡c-)片段与金原子配位,以提供更强的金-碳键,提高配合物的稳定性。其中,酰胺(包括芳酰胺和烷基酰胺,3a-3d)和酯基被用作在生物素的可修饰羧酸位置连接炔基配体的连接剂,目的是在生物相关条件下释放金。
[0081]
(3)在炔基金(i)配合物的结构中引入水溶性膦配体pta(配合物3b和3c)可以改变水溶性并降低细胞毒性,而引入亲脂性膦配体pph3(配合物3a和3d)可以增加膜溶解度并影响金配合物的药理特性。
[0082]
最终设计出生物素化金(i)配合物3a-3d,结构如下所示:
[0083][0084]
需要说明的是,金(i)表示金的化合价为+1。
[0085]
同时本发明也公开了上述配合物的制备方法,以及该配合物或其可药用的盐在制备抗肿瘤药物和放射增敏剂中的应用。
[0086]
本发明通过一系列实验对本发明提出的配合物,尤其是配合物3a的抗肿瘤活性和放射增敏效果进行了评价,并对其抗肿瘤机制和放射增敏机制进行了探究,结果发现,配合
物3a不仅能特异靶向肿瘤细胞,且能抑制肿瘤细胞增殖,最重要的是,配合物3a可通过靶向抑制trxr干预氧化还原稳态,并诱导肿瘤细胞铁死亡以增强辐射敏感性,且选择性高,直观地验证了本发明生物素化金(i)配合物在制备抗肿瘤药物和放射增敏剂中的应用。
[0087]
下面对本发明的实验过程及实验结果进行详细说明。
[0088]
实施例1具有肿瘤靶向的生物素化金(i)配合物的制备
[0089]
1、实验方法
[0090]
(1)中间体的制备
[0091]
中间体2a:称取d-生物素(8mg,0.03mmol)、4-乙炔基苯胺(35mg,0.3mmol)和o-(苯并三唑-1-基)-n,n,n',n'-四甲基铀六氟磷酸盐(hbtu,23mg)溶于1mldmf溶剂中,滴加三乙胺(0.12ml,0.06mmol),避光,在氮气保护下室温搅拌过夜。反应液用水(50ml)稀释后,乙酸乙酯萃取三次,合并有机层并洗涤,然后用无水硫酸钠干燥,过滤,并在减压下浓缩。将残余物通过硅胶柱色谱纯化(流动相:dcm∶meoh=10∶1),得到(n-(4-乙炔基苯基)-5-((3as,4s,6ar)-2-氧六氢-1h-噻吩并[3,4-d]咪唑-4-yl)戊酰胺)(中间体2a,6.07mg,收率59%)。
[0092]
中间体2b:称取1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐(edci)(0.408mg,2.12mmol)、d-生物素(0.400g,1.63mmol)和炔丙基胺(0.108g,1.96mmol)溶于乙腈:甲醇(3∶1,16ml)中,室温条件下搅拌反应6.5小时后浓缩。用甲醇复溶后,将溶液通过硅藻土过滤。将滤液真空浓缩,并通过硅胶色谱纯化(流动相:dcm∶meoh=10∶1),得到白色固体3-(d-生物素酰胺基)-1-丙炔(中间体2b,0.386g,收率84%)。
[0093]
中间体2c:称取d-生物素(1.0g,4.09mmol)、溴丙炔(0.52ml,4.58mmol)和k2co3(1.13g,8.18mmol)溶于dmf(30ml)溶液中。反应混合物在室温搅拌过夜后过滤。滤液用水稀释,并用乙酸乙酯萃取3次。合并的有机层用盐水洗涤,经无水硫酸钠(na2so4)干燥并蒸发。通过硅胶柱色谱纯化(流动相:ch2cl2∶ch3oh=30∶1),得到白色固体(乙炔基5-((3as,4s,6ar)-2-氧六氢-1h-噻吩并[3,4-d]咪唑-4-yl)戊酸酯)(中间体2c,1.06g,收率92%)。
[0094]
(2)生物素化金(i)配合物3a-3d的制备
[0095]
配合物3a:使用圆底烧瓶将中间体2a(50mg,0.14mmol)、koh(16mg,0.28mmol)和(三苯基磷)氯化金(i)(ph3paucl,72mg,0.14mmol)溶解在甲醇(meoh)中,避光,氮气保护下室温搅拌1小时。反应结束后,过滤反应混合物,用甲醇洗涤,得到白色固体三苯基膦基-[n-(4-乙炔基苯基)-5-((3as,4s,6ar)-2-氧六氢-1h-硫[3,4-d]咪唑-4-基)戊酰胺]金(i)(配合物3a,收率60%)。
[0096]
配合物3b:使用圆底烧瓶将中间体2a(50mg,0.14mmol)、koh(16.3mg,0.29mmol)和(1,3,5-三氮杂-7-磷三环[3.3.1.13,7]癸烷-κp7)-氯化金(i)(aupta,56.7mg,0.14mmol)溶解在甲醇(meoh)中,避光,氮气保护下室温搅拌1小时。反应结束后,过滤反应混合物,用甲醇洗涤,得到黄色固体(1,3,5-三氮杂-7-磷三环[3.3.1.13,7]癸烷-κp7)-n-(4-乙炔基苯基)-5-((3as,4s,6ar)-2-氧六氢-1h-噻吩并[3,4-d]咪唑-4-基)戊酰胺金(i)(配合物3b,收率55.4%)。
[0097]
配合物3c:使用圆底烧瓶将中间体2b(7.2mg,0.02mmol)、koh(2.8mg,0.05mmol)和aupta(10.0mg,0.02mmol)溶解在无水甲醇(meoh)中,避光,氮气保护下室温搅拌1小时。反应结束后,过滤反应混合物,用甲醇洗涤,并通过硅胶色谱纯化(流动相:dcm∶meoh=10∶1),得到白色固体(1,3,5-三氮杂-7-磷三环[3.3.1.13,7]癸烷-κp7)-5-((3as,4s,6ar)-2-氧
六氢-1h-噻吩并[3,4-d]咪唑-4-基)-n-(丙-2-基-1-基)戊酰胺金(i)(配合物3c,收率57.3%)。
[0098]
配合物3d:称取中间体2b(100mg,0.35mmol)、k2co3(97mg,0.7mmol)和ph3paucl(175mg,0.35mmol)溶解在10ml无水dmf中,避光,氮气保护下室温搅拌,tlc检测反应完全后,加水终止反应并用乙酸乙酯萃取3次。合并乙酸乙酯层并用无水na2so4干燥,过滤并蒸发。粗产物通过rp-hplc进一步纯化(固定相:c18;流动相:ch3cn/h2o),得到白色固体三苯基膦基-[5-(((3as,4s,6ar)-2-氧六氢-1h-噻吩并[3,4d]咪唑-4-基]-n-(丙-2-炔-1-基)戊酰胺]金(i)(配合物3d,收率25.3%)。
[0099]
(3)生物素化其他金属配合物3e-3h的制备:
[0100]
在合成上述四种生物素化金(i)配合物(3a-3d)的基础上,本发明还引入了其他过渡金属(例如含有二茂铁和羰基钴)作为生物活性对照,本实施例制备了配合物3e、3f、3g和3h。
[0101]
配合物3e:称取中间体2a(50mg,0.14mmol)和八羰基二钴(49.8mg,0.14mmol)溶解在10ml无水thf中,冰浴下搅拌,tlc检测反应完全后。加入1g硅胶,并将混合物蒸发至干后,并通过硅胶色谱纯化(流动相:dcm∶meoh=15∶1),得到红棕色固体[n-(4-乙炔基苯基)-5-((3as,4s,6ar)-2-氧六氢-1h-噻吩并[3,4-d]咪唑-4-基)戊酰胺]六羰基二钴(配合物3e,收率56%)。
[0102]
配合物3f:称取中间体2b(31.3mg,0.11mmol)和八羰基二钴(37.9mg,0.11mmol)溶解在10ml无水thf中,冰浴下搅拌,tlc检测反应完全后,加入1g硅胶,并将混合物蒸发至干后,并通过硅胶色谱纯化(流动相:dcm∶meoh=15∶1),得到红棕色固体[5-(((3as,4s,6ar)-2-氧六氢-1h-硫代[3,4-d]咪唑-4-基]-n-(丙-2-炔-1-基)戊酰胺]六羰基二钴(配合物3f,收率44.2%)。
[0103]
配合物3g:称取中间体2c(30.5mg,0.10mmol)和八羰基二钴(37.0mg,0.10mmol)溶解在10ml无水thf中,冰浴下搅拌,tlc检测反应完全后,加入1g硅胶,并将混合物蒸发至干后,并通过硅胶色谱纯化(流动相:dcm∶meoh=15∶1),得到红棕色固体[6-((3as,4s,6ar)-2-氧六氢-1h-噻吩并[3,4-d]咪唑-4-基]戊酸酯]的丙-2-炔-1-基]六羰基二钴(配合物3f,收率63.5%)。
[0104]
配合物3h:称取d-生物素(50mg,0.20mmol)溶解在10mldmf中,并依次添加4-二甲基氨基吡啶(dmap)(2.5mg,0.02mmol)、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺edci(43.0mg,0.22mmol)。搅拌10分钟后,将溶有二茂铁甲醇(44.0mg,0.20mmol)的dmf(5ml)溶液加入到反应混合物中。将反应混合物在室温搅拌过夜,然后过滤。滤液用水稀释,并用乙酸乙酯萃取3次。合并的有机层用盐水洗涤,经无水na2so4干燥并蒸发。通过硅胶色谱纯化(流动相:dcm∶meoh=30∶1),得到黄色固体5-(2-氧代六氢-1h-噻吩并[3,4-d]咪唑-4-基)戊酸二茂铁甲酯(配合物3h,收率45.5%)。
[0105]
2、表征和纯度鉴定
[0106]
对所上述制得的配合物3a-3h的结构和纯度等进行表征和鉴定。
[0107]
(1)核磁共振与质谱
[0108]
配合物3a:图1为配合物3a的核磁共振谱图。1h nmr(500mhz,dmso-d6)δ10.03(s,1h,-co-nh-),7.73-7.49(m,17h,-arh),7.23(d,j=8.4hz,2h,-arh),6.47(s,1h,-nh-),
6.39(s,1h,-nh-),4.35-4.25(m,1h),4.14(s,1h),3.15-3.07(m,1h),2.83(dd,j=12.3,4.9hz,1h),2.58(d,j=12.4hz,1h),2.31(t,j=6.8hz,2h),1.64-1.33(m,6h)。
[0109]
13
c nmr(126mhz,dmso)δ=171.63(-nh-co-nh-),163.18(-co-nh-),138.47,134.38,134.27,132.32,132.19,130.09,130.01,120.02,119.28,61.50,59.66,55.85,49.33,36.70,28.69,28.55,25。
[0110]
esi-ms[m/z]:802.2[m+h]
+
。
[0111]
配合物3b:图2为配合物3b的核磁共振谱图。1h nmr(500mhz,dmso-d6)δ9.90(s,1h,-co-nh-),7.49(d,j=8.4hz,2h,-arh),7.16(d,j=8.3hz,2h,-arh),6.41(d,j=37.6hz,2h,-nh-co-nh-),4.52(d,j=12.7hz,3h,-p-ch2-n,-ch-),4.39-4.26(m,11h,-p-ch2-n),4.14(s,1h,-ch-),3.12(t,j=9.2hz,1h),2.82(dd,j=12.4,4.9hz,1h),2.59(t,j=10.4hz,1h),2.29(t,j=7.1hz,2h),1.66-1.20(m,6h)。
[0112]
13
c nmr(126mhz,dmso)δ171.54(-nh-co-nh-),163.16(-co-nh-),138.28(-arc),132.11(-arc),120.27(-arc),119.22(-arc),72.33,72.26,61.49,59.65,55.85,51.26,51.11,40.57,36.69,28.69,28.54,25.54。
[0113]
esi-ms[m/z]:697.4[m+h]
+
。
[0114]
配合物3c:图3为配合物3c的核磁共振谱图。1h nmr(500mhz,dmso-d6)δ7.99(s,1h,-co-nh-),6.39(d,j=26.8hz,2h,-nh-co-nh-),4.51(d,j=12.6hz,3h,-p-ch2-n,-ch-),4.42-4.19(m,10h,-p-ch2-n),4.14(s,1h,-ch-),3.77(d,j=4.4hz,2h),3.10(s,1h),2.83(dd,j=12.2,4.4hz,1h),2.58(d,j=12.4hz,1h),2.04(t,j=6.8hz,2h),1.66-1.21(m,6h)。
[0115]
13
c nmr(500mhz,dmso)δ171.79(-nh-co-nh-),163.15(conh),72.33,72.27,61.48,59.66,55.84,51.26,51.10,35.38,29.54,28.65,28.45,25.63。
[0116]
esi-ms[m/z]:[m+h]
+
635.2,[m+na]
+
657.1。
[0117]
配合物3d:图4为配合物3d的核磁共振谱图。1h nmr(500mhz,dmso-d6)δ8.06(s,1h,-co-nh-),7.73-7.40(m,15h,arh),6.41(s,1h,-nh-),6.34(s,1h-nh-),4.28(s,1h),4.13(s,1h),3.85(d,j=4.8hz,2h,-nh-ch2-),3.10(s,2h),2.91-2.72(m,2h,-ch2-co-o-),2.56(d,j=12.4hz,1h),2.07(t,j=7.0hz,2h),1.41(d,j=96.9hz,6h)。
[0118]
13
c nmr(500mhz,dmso)δ171.85(-nh-co-nh-),163.14(-co-nh-),134.36,134.25,132.39,130.11,130.02,129.59,61.47,59.62,55.87,55.39,35.38,29.55,28.68,28.47,25.67。
[0119]
esi-ms[m/z]:740.1[m+h]
+
。
[0120]
配合物3e:图5为配合物3e的核磁共振谱图。1h nmr(500mhz,dmso-d6)δ10.08(s,1h,-co-nh-ph),7.63(d,j=8.1hz,2h),7.49(d,j=8.2hz,2h),7.11(s,1h,-c≡ch),6.45(s,1h,-nh-),6.37(s,1h,-nh-),4.31(s,1h,-nh-ch-),4.15(s,1h,-nh-ch-),3.13(s,1h),2.83(dd,j=12.3,4.7hz,1h),2.58(d,j=12.4hz,1h),2.32(t,j=6.7hz,2h,-ch2-co-nh-),1.43(ddd,j=49.3,40.4,38.4hz,6h)。
[0121]
13
c nmr(126mhz,dmso)δ200.26(co-co),171.85(-nh-co-nh-),163.22(-co-nh-),139.99,131.15,131.00,119.92,90.85,74.53,61.51,59.67,55.86,36.72,28.67,28.56,25.56。
[0122]
esi-ms[m/z]:679.2[m+cl]-。
[0123]
配合物3f:图6为配合物3f的核磁共振谱图。1h nmr(500mhz,dmso-d6)8.47(t,1h,-co-nh-),6.64(s,1h,-c≡ch),6.44(s,1h,-nh-),6.35(s,1h,-nh-),4.45(d,2h,-nh-ch-),4.31(at,1h),4.12(at,1h),3.10(m,1h),2.82(dd,1h),2.58(d,1h),2.11(t,2h,-ch2-co-nh-),1.68-1.24(m,6h)。
[0124]
13
c nmr(126mhz,dmso)δ200.42(co-co),172.22(-nh-co-nh-),163.19(-co-nh-),96.15,73.43,61.49,59.65,55.87,40.93,35.47,28.82,28.56,25.38。
[0125]
esi-ms[m/z]:618.0[m+k]
+
。
[0126]
配合物3g:图7为配合物3g的核磁共振谱图。1h nmr(500mhz,chloroform-d)δ5.54(s,1h,-nh-),5.08(s,1h,-nh-),4.70(d,j=2.4hz,2h,-o-ch2-c≡ch),4.62-4.46(m,1h),4.40-4.26(m,1h),3.18(dd,j=4.7,1.8hz,1h),2.95(dd,j=12.8,5.0hz,1h),2.77(d,j=12.8hz,1h),2.51(t,j=2.4hz,1h,-c≡ch),2.42(t,j=7.4hz,2h,-ch2-co-),1.81-1.70(m,3h),1.66(d,j=5.7hz,1h),1.48(td,j=15.4,8.0hz,2h)。
[0127]
13
c nmr(126mhz,cdcl3)δ199.13(co-co),173.19(-nh-co-nh-),163.42(-ch2-co-o),88.89,71.80,64.76,61.94,60.11,55.30,40.55,33.55,29.69,28.38,28.28,24.53。
[0128]
esi-ms[m/z]:618.0[m+cl]-。
[0129]
配合物3h:图8为配合物3h的核磁共振谱图。1h nmr(500mhz,dmso-d6)δ6.43(s,1h,-nh-),6.37(s,1h,-nh-),4.86(s,2h),4.29(dd,j=10.1,4.5hz,3h,-ch-;fe(c5h4)),4.23-4.15(m,7h,fe-c5h5;fe(c5h4)),4.14-4.09(m,1h),3.11-3.02(m,1h),2.90-2.82(m,1h),2.81-2.72(m,1h),2.57(d,j=12.4hz,1h),2.27(t,j=7.4hz,2h),1.61-1.30(m,6h)。
[0130]
13
c nmr(500mhz,dmso)δ173.04(-nh-co-nh-),163.13(-co-nh-),162.76,81.89,69.81,68.88,62.50,61.46,59.62,55.79,36.25,33.73,31.24,28.42,24.99。
[0131]
esi-ms[m/z]:445.4[m+h]
+
。
[0132]
(2)高效液相色谱(hplc)分析
[0133]
使用hplc检测所得配合物3a-3h的纯度,实验条件为:色谱柱:inertex c18(250mm
×
4.6mm
×
5μm),流动相为甲醇-水或乙腈-水(5%-95%至100%-0,v/v),波长:254nm。速率:0.5或0.8ml/min,温度:25℃。结果如图9中的a-h图所示,hplc检测所得配合物3a-3h的纯度均大于95%。
[0134]
实施例2评价配合物3a-3h的体外选择性和抗肿瘤活性
[0135]
1、细胞摄取能力的检测
[0136]
为了评估实施例1制备的配合物3a在肿瘤细胞中的摄取情况,以金诺芬(auranofin)处理为对照,配合物3a处理作为实验组,生物素预孵育2h后,配合物3a处理作为竞争测试组,通过hela和h8细胞孵育12h后,使用电感耦合等离子体质谱(icp-ms)测定细胞内au浓度。
[0137]
结果如图10所示,在hela细胞中,配合物3a(910
±
79ng/106个细胞)的积累是金诺芬(441
±
60ng/106个细胞)的2.0倍,这可能与引入的生物素组有关。此外,观察到生物素预处理的hela细胞减少了对3a的摄取(696
±
27ng/106个细胞),表明hela细胞对3a的摄取由部分生物素受体介导。另外,在相同条件下,h8细胞配合物3a的摄取能力明显低于hela细胞。
[0138]
以上结果表明,本技术配合物中引入的生物素结构可以增加细胞对金(i)配合物的吸收。同时,生物素化的金(i)配合物3a对各种细胞系中的癌细胞显示出选择性。
[0139]
2、配合物对肿瘤细胞的抗增殖活性和选择性的检测
[0140]
取人宫颈癌细胞(hela和siha)和人宫颈上皮细胞(h8)在37℃、5%co2条件下培养于含10%的胎牛血清和1%青霉素-链霉素的dmem培养基中。根据细胞类型,将前述状态良好的细胞接种到96孔板中,每孔约1200~3000个细胞,并使其附着过夜。第二天加入不同的化合物(均用dmf作溶剂),对照组用0.1%dmf处理,处理过的细胞培养72h后,通过mtt法检测所有生物素金属配合物对宫颈癌细胞(siha和hela)和正常宫颈细胞(h8)的抗增殖活性。
[0141]
结果见表1,对于hela细胞,所有生物素化金(i)配合物均有一定的增殖抑制作用,其中配合物3a的细胞毒性与阳性对照药物金诺芬相当,并且优于顺铂。然而,钴和二茂铁配合物没有显示明显的抗增殖活性。对于siha细胞,配合物3a的抗增殖活性仍与金诺芬相当,且优于顺铂。另外,根据表1可知,配合物3a对hela细胞具有更高的选择性(si值为3.0),显著优于金诺芬(si值为1.6)。
[0142]
表1配合物对hela、siha与h8细胞的抗增殖活性和选择性
[0143][0144]
[a]si=ic
50
(正常细胞)/ic
50
(肿瘤细胞)
[0145]
实施例3配合物3a的抗肿瘤机制研究
[0146]
基于实施例2可知,本技术中的配合物3a表现出优异的抗肿瘤活性和肿瘤细胞选择性,因此,继续对配合物3a的抗肿瘤机制进行一系列研究。
[0147]
1、配合物3a抑制trxr活性
[0148]
(1)体外纯化的trxr活性的测定
[0149]
dmso或不同浓度3a和大鼠肝脏trxr溶解在50
μ
l反应缓冲液中。向每个孔中添加225μl反应混合物。通过添加25μl 20mm dtnb溶液开始反应。适当混合后,用微孔板读取器记录405nm处的吸收数据。tnb浓度随时间的增加呈线性趋势(r2》0.99),酶活性计算为其斜
率(每30秒增加的吸光度)。
[0150]
结果如图11中的a图所示,纯化的trxr活性测定表明,配合物3a以剂量依赖性方式抑制trxr。
[0151]
(2)细胞trxr活性的测定
[0152]
培养hela细胞生长至70~80%的丰度时,用不同浓度的3a处理并孵育72h。小心弃去培养基,用pbs洗涤两次。在冰浴上通过ripa缓冲液处理提取总细胞蛋白。使用bradford程序量化。根据trxr活性检测试剂盒测定细胞中trxr的活性。
[0153]
结果如图11中的b图所示,配合物3a在处理72h后显示出对细胞trxr活性抑制的作用(ic50=0.74μm),且随着3a浓度的增大,活性抑制越明显。该结果进一步支持配合物3a的抗肿瘤活性源于其对trxr的抑制作用的观点。
[0154]
(3)荧光检测细胞内trxr活性
[0155]
为了进一步验证3a可以直接作用于细胞内trxr的想法,利用报告的探针来可视化hela细胞中的trxr。将接种在6孔板中的细胞用3a(2μm)处理24h,并在新鲜培养基中用trfs green(10μm)再孵育0.5h,然后用pbs洗涤两次,荧光显微镜下拍照获得荧光图像。
[0156]
结果如图11中的c图所示,仅0.5h的3a(2μm)处理后,细胞内trxr活性几乎消除。
[0157]
(4)western blot检测细胞内trxr的表达水平
[0158]
将hela细胞接种到直径为10cm的组织培养皿(10ml/孔)中,在37℃、5%co2的潮湿环境中培养24h,然后用3a处理24h,小心弃去培养基,用冷pbs洗涤三次。在冰浴中用ripa缓冲液裂解,离心收集上清液并通过bca定量。通过12.5%sds-page凝胶分离样品,tris-甘氨酸作为涌动液。分离结束后,将蛋白质印迹到pvdf膜上,并通过各种特异性抗体检测。然后将它们洗涤并与抗兔或抗鼠一起孵育2小时。用化学发光法(ecl)检测蛋白条带。
[0159]
结果如图11中的d图所示,尽管细胞内的trxr活性被3a显著抑制,但细胞内trxr蛋白水平是不变的。
[0160]
2、配合物3a不与dna结合
[0161]
通过紫外-可见光谱法和圆二色光谱法检测3a和小牛胸腺dna(ct-dna)的结合作用。所有光谱分析均在pbs缓冲液(10mm,ph=7.2)中进行。将ct-dna(1.580mm)储备液储存在4℃的冰箱中,使用前保存5天以下。将3a溶解在dmf中并混合成20mm储备溶液,使用pbs缓冲液将其稀释至相应浓度,以进行后续光谱实验。
[0162]
紫外-可见光谱法:将3a储备液用pbs缓冲液稀释1000倍,得到2.0
×
10-5
m溶液(3ml),然后逐渐加入ct-dna储备液,混合溶液充分反应时,记录uv-vis吸收光谱。
[0163]
圆二色光谱法:将3a添加到ct dna(1.0x 10-5m)溶液中,并在37℃下培养5分钟。3a的最终浓度为0,0.02,0.04,0.08,0.1,0.2,0.4,0.6,1.0,2.0
×
10-5
m,每个样品用cd光谱仪记录200-320nm范围内的数据。结果如图12的a-c图所示,dna的吸收和显色以及构象没有随着3a浓度的增加而发生显著变化,由此说明配合物3a不与dna结合。
[0164]
3、配合物3a显著增加hela细胞内ros水平
[0165]
(1)荧光探针检测3a对处理hela细胞中ros的影响
[0166]
荧光dcfh-da本身没有荧光,可以自由穿过细胞膜。进入细胞内后,可以被细胞内的酯酶水解生成dcfh,而dcfh不会通透细胞膜,因此探针很容易被积聚在细胞内。细胞内的活性氧能够氧化无荧光的dcfh生成有绿色荧光的dcf。绿色荧光强度与活性氧的水平成正
比。将hela细胞分别与不同浓度的配合物3a或顺铂(cisplatin)孵育12h,然后用pbs洗涤并在37℃、黑暗中用10μm的dcfh-da处理20分钟。通过荧光显微镜检测荧光的增加。
[0167]
结果如图13中的a图所示,与3a一起孵育的hela细胞以剂量依赖性方式显示出强烈的荧光信号,表明添加3a产生了大量的细胞内ros。
[0168]
(2)流式细胞术测定不同处理方法对hela细胞中ros水平的影响
[0169]
hela细胞经4mm n-乙酰半胱氨酸(nac)预处理1h或不预处理后,加入2μm的配合物3a孵育6h,然后用pbs洗涤细胞,用5μm dcfh-da在培养基中培养30min,再用pbs洗涤两次。通过流式细胞仪检测荧光强度,计数10000个细胞。使用flowjo 10.7.1进行分析。
[0170]
结果如图13中的c图所示,nac预处理1h后,3a的荧光强度显着降低至与对照组无差异的水平。
[0171]
4、配合物3a导致线粒体功能受损
[0172]
继续研究配合物3a对线粒体的影响。主要考察指标为不同浓度3a作用后线粒体膜电位(mmp)水平的变化。将hela细胞接种于24孔板中,用不同浓度复合物3a或dmf或顺铂处理24h,然后用jc-1染料(5μm)染色30分钟。通过荧光显微镜拍摄。其结果如图13中的b图所示,与dmf处理组相比,3a处理组的绿色(单体)荧光增加,说明配合物3a对线粒体的破坏增加。
[0173]
此外,hela细胞经4mm n-乙酰半胱氨酸(nac)预处理1h或不预处理,再用pbs洗涤两次,并与3a(2μm)孵育0.5小时。收集用指定实验条件处理的细胞,然后在37℃的条件下与jc-1染料(5μm)在黑暗中孵育20分钟。之后用冷冻pbs冲洗细胞并通过流式细胞仪分析,计数10000个细胞。使用flowjo 10.7.1进行分析。其结果如图13中的d图所示,nac可以显著抑制ros诱导的膜电位(mmp)水平。
[0174]
5、配合物3a对hela细胞凋亡和细胞周期的影响
[0175]
(1)fitc和碘化丙啶双染法检测3a对hela细胞凋亡的影响
[0176]
将hela细胞接种在6孔板中,当细胞密度达70~80%时,分别用3a(2μm)和顺铂(2μm)处理细胞48h,然后根据试剂盒说明书,用流式细胞仪检测细胞凋亡情况,其结果如图14中的a图所示,与用顺铂处理的细胞相比,配合物3a(2μm)处理48h后显著增加了细胞凋亡水平。
[0177]
(2)检测凋亡通路蛋白bax/bcl-2的表达
[0178]
western blot检测了凋亡信号通路中bax/bcl-2的表达水平,其结果如图14中的b图所示,用3a(2μm)和顺铂(2μm)处理细胞48h后,3a降低了抗凋亡蛋白bcl-2的表达,增加了促凋亡蛋白bax水平。
[0179]
(3)碘化丙啶染色法检测3a对hela细胞周期的影响
[0180]
hela细胞悬液均匀的加入到6孔板中,贴壁后,设置对照组(dmf)与不同浓度的3a组,置于培养箱中共孵育24h。用pbs清洗一遍,用不含edta的胰酶消化,离心,弃上清。加入预冷的70%乙醇置于冰箱对细胞进行固定。次日将已经固定好的细胞离心、洗涤,用pbs重悬,加入rnasea酶,在37℃水浴锅中保持30min,之后加入已经配好的染色剂pi,于4℃冰箱中避光作用15min,上机检测。
[0181]
结果如图14中的c图所示,随着3a浓度的增加,g2/m期细胞所占比例明显升高,表明配合物3a能影响hela细胞的细胞周期,并将细胞阻滞在放疗敏感的g2/m期。
[0182]
6、配合物3a的抗肿瘤细胞增殖活性(mtt)实验
[0183]
对配合物3a在宫颈癌细胞hela中进行了体外细胞毒性实验,在96孔细胞培养板中接种细胞(2000个/孔)并培养过夜,加入不同浓度的3a继续培养72h,dmf作空白对照,并用或不用各种浓度的nac预处理1h,每孔加入0.5%的mtt试液孵育4h,小心弃去培养液,每孔加入200μldmso溶解沉淀,在摇床上低速振摇10min,用酶标仪在490nm处测量每孔的吸收值。
[0184]
其体外细胞毒性结果如图15所示。nac(≥4mm)几乎完全消除了复合物3a诱导的细胞毒性。
[0185]
因此,本实施例所有实验数据表明配合物3a可以有效地在hela细胞中产生ros,导致线粒体功能受损、细胞周期停滞和细胞凋亡,这些都与3a的细胞毒性有关。
[0186]
实施例4配合物3a的体外放疗增敏效果
[0187]
(1)cck8检测不同浓度3a和不同剂量x射线处理后的细胞活力
[0188]
将hela细胞接种到96孔板中,并使其贴壁过夜。hela细胞与不同浓度3a孵育24h,然后用不同剂量x射线照射,37℃孵育24h。最后,每孔加入10微升cck8试剂,孵育半小时,用酶标仪在450nm处检测吸光度。
[0189]
结果如图16中的a图所示,3a联合x射线照射后hela细胞的生长抑制呈剂量依赖性增加,而单独的x射线的细胞毒性显示出最小的毒性。
[0190]
为了进一步评估x射线和3a之间的相互作用,继续进行了组合指数分析。结果如图16中的b图所示。组合指数图中的数据点位于定义加性效应的线下方,表明不同剂量的3a和x射线联合治疗具有协同效应。
[0191]
(2)克隆生成试验
[0192]
将hela和h8细胞分别接种在6孔板中,用3a处理或不处理24小时后,并分别暴露于x射线。在37℃、5%co2中培养10至14天,用pbs冲洗细胞3次,在甲醇中固定,然后进行结晶紫染色。菌落数量用image-j进行量化。
[0193]
结果如图16中的c-d图所示,3a和x射线联合治疗对hela细胞有协同抑制作用,存活率降至7.9%。更重要的是,3a对hela细胞的作用远远好于正常h8细胞(58.3%),超过7倍,这再次证明3a对肿瘤细胞辐射增敏的选择性。
[0194]
实施例4配合物3a的放疗增敏机制研究
[0195]
(1)cck8检测不同浓度3a和x射线处理后的细胞活力
[0196]
将hela细胞以每孔1000个细胞的数量接种到96孔板,培养24h后先加入浓度为4mm的nac培养1h,再加入不同浓度的3a进行培养。24h后进行2gy放疗。再培养24h,加10μl cck8试剂,0.5h后于450nm处测od值。
[0197]
结果如图17中的a图所示,在x射线(2gy)和3a处理组中,用ros清除剂nac预处理的hela细胞活力显著提高,当3a浓度为2.5μm或更低时,细胞活力没有明显下降。
[0198]
(2)western blot检测不同处理后hela细胞中γ-h2ax蛋白的表达水平
[0199]
ros介导的dna损伤的诱导是辐射诱导的细胞死亡的重要机制,于是通过量化γ-h2ax的磷酸化状态来检测双链dna断裂情况。将hela细胞接种于6孔板,先加入4mm的nac培养1h,再用3a(2.5μm)处理或不处理24h后,分别暴露于由x射线直线加速器产生的0gy和2gy x射线下培养24h。小心弃去培养基,用冷pbs洗涤三次。在冰浴中用ripa缓冲液裂解,离心收
集上清液并通过bca定量。通过12.5%sds-page凝胶分离样品,tris-甘氨酸作为电泳液。分离结束后,将蛋白质印迹到pvdf膜上,并通过各种特异性抗体检测。然后,将它们洗涤并与抗兔或抗鼠一起孵育2h。用化学发光法(ecl)检测蛋白条带。
[0200]
结果如图17中的b图所示,辐射(2gy)或3a(2.5μm)单独增加了γ-h2ax的表达水平,联合治疗显示出加性效应,而nac预处理抑制了这种效应。
[0201]
(3)cck8检测铁死亡抑制剂ferrostatin-1的影响
[0202]
将hela细胞接种于96孔板,培养24h后,先加入不同浓度的铁死亡抑制剂ferrostatin-1培养1h,再加入不同浓度的3a进行培养。24h后进行0gy或2gy放疗,再培养24h,加10μl cck8试剂,0.5h后于450nm处测od值。
[0203]
结果如图17中的c-d图所示,铁死亡抑制剂ferrostatin-1预处理在一定程度上增加了暴露于3a、ir联合治疗的hela细胞的存活率;重要的是,仅当3a浓度为1μm时,细胞存活的恢复比高剂量时更明显。这意味着与ros积累相比,3a引起的铁死亡对放疗致敏的影响相对较小。
[0204]
(4)western blot检测不同处理后hela细胞中gpx-4蛋白的表达水平
[0205]
将hela细胞接种于六孔板,先加入浓度为1μm的ferrostatin-1,再用3a(1μm)处理或不处理24h后,并分别暴露于由x射线直线加速器产生的0和2gy x射线后,于培养箱中培养24h。用冷pbs洗涤三次。在冰浴中用ripa缓冲液裂解,然后离心,收集上清液并通过bca定量。通过12.5%sds-page凝胶分离样品,tris-甘氨酸作为电泳液。分离结束后,将蛋白质印迹到pvdf膜上,并通过各种特异性抗体检测。然后,将它们洗涤并与抗兔或抗鼠一起孵育2h。用化学发光法(ecl)检测蛋白条带。
[0206]
结果如图17中的e图所示,在放射治疗中,尽管会发生铁死亡,但也会诱导gpx-4表达增加,抵抗放射治疗。然而,放射联合3a治疗有效地改善了这种情况,显着抑制了gpx-4的表达并促进了肿瘤细胞中的铁死亡。此外,单独施用3a也显著抑制了gpx-4的表达并促进铁死亡。
[0207]
综上,本实施例实验结果表明,配合物3a的放射增敏作用是通过ros积累和铁死亡引起的氧化还原失衡介导的。
[0208]
实施例5本发明所合成的配合物3a的体内抗肿瘤实验
[0209]
在内皮细胞中表达增强型绿色荧光蛋白(egfp)的转基因斑马鱼tg(fli-1:egfp)购自南京伊泽林卡生物技术有限公司,斑马鱼被饲养在循环水产养殖系统中,在标准实验室条件下,标准昼夜光照周期为28.0
±
1.0℃。对照组用0.1%dmf,治疗组单独暴露于3a(1.25μm)和顺铂(1μm),照射剂量为10gy,顺铂结合ir(电离辐射),3a结合ir。所有实验均在胚胎移植成功后第2天进行:受精后2天(dpf),因为胚胎移植后1天需要观察。
[0210]
(1)放疗和3a对斑马鱼胚胎发育的影响
[0211]
在受精后24h,用不同的处理方法处理去除绒毛的斑马鱼胚胎。将胚胎于0.03mg/ml三卡因中镇静,用荧光显微镜对血管缺陷进行活体成像研究。计算孵化率和存活率,并进行统计分析。
[0212]
结果如图18中的a-g图所示,在单独放疗、3a+ir联合治疗或顺铂+ir联合治疗36h的胚胎中未发现胚胎畸形和对血管生成的影响。与dmf处理的胚胎(对照组)相比,药物组在有无放射治疗的情况下孵化率或存活率没有显著差异。
[0213]
(2)3a抑制ir对斑马鱼胚胎内生长的人类宫颈癌细胞的转移
[0214]
hela细胞被接种在6孔板中,并生长至80%汇合。根据制造商的方案,用cm-dil(2μg/ml,溶解于dmso中,并在hbss中稀释)标记培养细胞。将100至200个细胞移植到胚胎卵黄囊中心,即绒毛膜斑马鱼胚胎(24hpf)。注射后,胚胎在28℃下保持1小时,然后在31℃下孵化。细胞移植后,用荧光显微镜连续观察胚胎。拍摄亮场和荧光图像,并测量肿瘤区域的荧光强度。所有治疗组的胚胎处理方法相同。
[0215]
结果如图19中的a-b图所示,所有胚胎肿瘤的荧光在治疗后显著降低,除顺铂组外,所有其他治疗组均能有效抑制肿瘤转移,3a联合放疗的效果最为明显。
[0216]
综上所述,本发明通过一系列实验对本发明制备方法制得的生物素化金(i)配合物尤其是配合物3a的抗肿瘤活性和放射增敏效果进行了评价,并对其抗肿瘤机制和放射增敏机制进行了探究,结果发现,配合物3a不仅能特异性靶向肿瘤细胞,且抑制肿瘤细胞增殖,还可通过靶向抑制trxr干预氧化还原稳态,并诱导铁死亡以增强辐射敏感,且择性高,具有良好的市场应用前景。
[0217]
尽管本发明的内容已经通过上述优选实施例作了详细介绍,但应当认识到上述的描述不应被认为是对本发明的限制。在本领域技术人员阅读了上述内容后,对于本发明的多种修改和替代都将是显而易见的。因此,本发明的保护范围应由所附的权利要求来限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1