一种利用高通量测序技术检测多基因位点突变的方法与流程
1.本发明属于基因测序技术领域,具体涉及一种利用高通量测序技术检测多基因位点突变的方法。
背景技术:
2.癌症是威胁人类健康和生命安全的头号杀手,随着人类基因组计划的完成和后基因组时代的到来,癌症的治疗方法已不仅仅局限于手术、放疗、化疗、中药治疗和生物治疗等,近年来兴起的靶向治疗已表现出明显优势并占据重要地位。
3.靶向治疗是肿瘤综合治疗中一个重要的治疗手段,是指以肿瘤细胞的标志性分子为靶点,研制出有效的阻断剂,干预细胞癌变的各个环节。靶向治疗与传统的放疗、化疗有明显的不同,主要针对肿瘤细胞起作用,对正常的细胞影响较小,所以在治疗的时候更加有针对性。
4.靶向治疗通过抑制肿瘤细胞增殖、干扰细胞周期、诱导肿瘤细胞分化、抑制肿瘤细胞转移、诱导肿瘤细胞凋亡,或者抑制肿瘤的血管生成等途径,达到抗肿瘤治疗的目的。所以靶向治疗的靶点包括肿瘤细胞生长因子受体、信号转导分子、细胞周期蛋白、细胞凋亡的调节因子、蛋白水解酶、血管内皮细胞生长因子等,药物直接作用在相应的靶点,起到抗肿瘤的作用。
5.常见的基因突变检测平台和技术包括:一代测序技术、变性高效液相色谱(dhplc)技术、荧光定量pcr技术、微滴式数字pcr技术、荧光原位杂交(fish)技术和第二代高通量测序(ngs)技术等。
6.近年来,液体活检的概念开始兴起,其检测对象从患者的组织变成体液样本(血液占大多数),在肿瘤的筛查和诊断,药物的疗效监测和患者预后判断等领域,都具备很高的价值。作为一种较好的肿瘤组织替代样本类型,血液样本中的ctdna(循环肿瘤dna)含量却很低,这就要求采用的检测技术需要具备极高的检测灵敏度。
7.上述基于pcr平台的检测方法,在检测时间、人力成本和检测灵敏度方面等具有一定优势,但以荧光定量pcr为例,根据目前主流的荧光pcr仪器检测通道数量,单管反应所能检测的靶基因数目很难突破4-6个,这也限制了基于pcr平台检测技术在涉及多靶点的复杂疾病上的应用。而作为“金标准”的一代测序,并不具备很高的检测灵敏度(约10%)。相较于一代测序,变性高效液相色谱技术的灵敏度略有提高,但其操作步骤复杂、周期长和极易造成污染的特点,也限制了其应用范围。荧光原位杂交技术可用于基因扩增、缺失和重排的检测,但同样面临着探针不能100%杂交、操作繁琐和耗时长等困境。
8.因此,本研究领域迫切需要开发一种高特异性、高灵敏度、高通量的基因检测方法。
9.第二代测序(next-generation sequencing,ngs)又称高通量测序(high-throughput sequencing),是基于pcr和基因芯片发展而来的dna测序技术。近年来,ngs技术的引入为探究疾病发生的分子机制提供了新的见解,并已经广泛应用于多种恶性肿瘤的
诊断、分型、预后危险度分层、治疗手段筛查、疗效评估和复发预测等方面。随着ngs技术的不断完善和规范化,其在疾病纵向监测和疾病评估中的作用越来越引起临床医生的关注。ngs技术又包括全基因组测序(whole genome sequencing,wgs)、全外显子测序(whole exon sequencing,wes)、转录组测序(rna-seq)和靶基因测序(targeted sequencing)等。靶向ngs,亦称为ngs panel,是临床上应用最广泛的基于ngs的工具,该技术通过靶向选择感兴趣的关键基因,通常对已知临床相关突变的“热点”区域进行测序,而不是对整个基因进行测序,是一种可最大限度地关注这些区域的高通量测序技术。因此,ngs panel需要密切掌握目标疾病的关键驱动基因、基因结构及其突变性质,以设计出针对某疾病中关键基因的特定panel。目前,ngs panel凭借其成本低、检测时间短、检测深度高等优点,已被广泛应用到癌症的诊断、预后危险度分层、靶向治疗方案选择、疗效评价及复发预测等方面,受到越来越多医生和患者的青睐。然而同时,不可忽略的是,ngs平台引入的系统误差,使我们难以检测到低频或罕见的变异。
10.因此,为了更好地实现对于低丰度变异的检测,同时又能够满足检测成本低、建库流程短的需求,就需要设计一种高效的文库结构和文库制备体系。
技术实现要素:
11.本发明的目的在于提供一种利用高通量测序技术检测多基因位点突变的方法,克服了现有技术的不足,采用特异性的引物和探针组合,抑制样品中野生型序列的扩增,同时富集低丰度突变型序列;通过高效的文库结构和制备体系,实现在高通量测序平台对待测样本中多个基因多个位点同时进行检测,其检测灵敏度可低至0.01%。
12.为解决上述问题,本发明所采取的技术方案如下:
13.一种利用高通量测序技术检测多基因位点突变的方法,包括以下步骤:
14.步骤一、末端修复:在dna聚合酶的作用下,对cfdna进行末端修复,使双链dna形成平末端,并分别在5’端进行磷酸化和3’端加da尾;
15.步骤二、接头连接:通过a-t连接的方式,将i5端接头序列连接至模版dna的5’端,所述i5端接头序列包括i5端通用引物结合靶点、i5端样本标签、i5端测序引物重叠序列、特异性随机标签序列和间隔序列;
16.步骤三、第一次pcr反应:使用特异性探针抑制野生型dna序列的扩增,使用i5端通用引物和i7端预扩增引物,预扩增突变型dna序列;
17.步骤四、第二次pcr反应:使用i5端通用引物、i7端特异性引物和i7端接头,进一步富集第一次pcr反应所得产物,所述i7端接头包括p7端测序芯片寡核苷酸簇互补序列、i7端样本标签、i7端测序引物重叠序列、i7端特异性引物互补序列的一部分;
18.步骤五、文库质控和测序分析。
19.进一步,步骤二中所述i5端接头序列的长度为79-88个碱基,其中,i5端通用引物结合靶点序列为20-29个碱基、i5端样本标签序列为8个随机碱基、i5端测序引物重叠序列为33个碱基、特异性随机标签序列为12个碱基以及间隔序列为6个碱基,所述i5端通用引物结合靶点序列可与p5端测序芯片寡核苷酸簇序列互补。
20.进一步,步骤三中所述特异性探针长度为35-50个碱基,其序列和野生型待测序列互补,包括blocker部分和reporter部分,所述特异性探针的tm值比i5端通用引物的tm值高
15-25℃。
21.进一步,步骤三中所述i5端通用引物用于扩增目标区域并与i5端接头上的通用引物结合靶点互补,其长度为15-25个碱基。
22.进一步,步骤三中所述i7端预扩增引物用于预扩增目标区域,其序列长度为15-25个碱基。
23.进一步,步骤四中所述i7端特异性引物的长度为15-25个碱基。
24.进一步,步骤四中所述i7端接头序列长度为58-62个碱基,其中,所述p7端测序芯片寡核苷酸簇互补序列为24个碱基、i7端样本标签序列为8个碱基、i7端测序引物重叠序列为20个碱基、与i7端特异性引物部分互补的序列为6-10个碱基。
25.进一步,所述方法包括三次磁珠纯化,其中第一次磁珠纯化用于对步骤二中连接接头后的产物进行纯化,第二次磁珠纯化用于对步骤三中第一次pcr反应的产物进行纯化,以及第三次磁珠纯化用于对步骤四中第二次pcr反应的产物进行纯化。
26.本发明与现有技术相比较,具有以下有益效果:
27.1、本发明在预扩增步骤中引入了用于抑制野生型dna序列扩增的特异性探针,在不对称扩增的条件下,设置合适的退火温度,使得探针先于引物与dna模版结合,i5端通用引物可与待测序列一条链进行结合,i7端预扩增引物可与待测序列的另一条链结合。对于突变型dna模版序列,在合适的延伸温度条件下,与靶序列结合的i5端通用引物不受探针影响,具有延伸能力;对于野生型序列,在设定的延伸温度条件下,与dna模版靶序列结合的i5端通用引物,其延伸能力被抑制。上述反应过程将突变模版dna进行富集和放大,从而提高了检测的灵敏度,其检测灵敏度可低至0.01%。
28.2、本发明在pcr反应之前引入了umi,该随机序列作为分子标签,是对原始样本目标区域基因组上的每一个片段都加上一段特有的标签序列,在后续pcr反应和测序过程中,来源相同原始模版的dna片段带有相同的umi,因此可对检测过程中随机产生的错误碱基进行校正,降低错误率,保证检测结果的准确性。
29.3、本发明对dna起始量的要求更低,因此更适合稀有样本和低丰度突变的检测;不需要采用价格昂贵的生物素标记的探针和链霉亲和素磁珠,且仅需很少的测序数据量,就能够获得很高的测序深度,因此可大幅降低检测成本;由于不需要进行过夜杂交,因此可将2天的实验流程缩短至当天完成;具有很强的检测未知基因融合变异的能力,进而可以发现更多未发表过的复杂基因突变。
附图说明
30.图1为本发明中探针结构示意图。
31.图2为本发明方法的流程示意图。
32.图3为本发明方法的文库片段分布示意图。
具体实施方式
33.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他
实施例,都属于本发明保护的范围。
34.实施例1
35.参见图1,本实施公开了一种特异性探针结构,探针从5’端到3’端依次为blocker和reporter部分。特异性探针的tm值比i5端通用引物的tm值高15-25℃。
36.特异性探针可以被硫代修饰、脱氧脲嘧啶修饰、脱氧次黄嘌呤修饰、2-甲氧基修饰、磷酸化修饰中的任意一种方式修饰,也可以是多种方式同时修饰。
37.其中,blocker部分的长度为15-20个碱基,包括人工引入的2-6个任意碱基序列,其它位点和野生型待测dna序列互补,blocker部分的5’端引入茎环结构,且茎环结构的长度为3-10个碱基,其序列和blocker其中一部分序列互补;reporter部分,其长度20-30个碱基,其序列和野生型待测序列互补。
38.参见图2,本实施例还公开了一种利用高通量测序技术检测多基因位点突变的方法,具体实施过程包括以下部分:
39.步骤一、末端修复:对cfdna进行末端修复,使双链dna形成平末端,并分别在5’端进行磷酸化和3’端加da尾。采用一步反应,可对极低投入的dna样本进行高效、快速的末端修复和da尾添加,同时保证更高的转化效率。
40.步骤二、接头连接:通过a-t连接的方式,将i5端接头序列连接至模版dna的5’端,用于添加i5端通用引物结合靶点、i5端样本标签、i5端测序引物重叠序列、特异性随机标签(umi)序列和间隔序列。本步骤通过连接接合,而非pcr放大的方式,可大幅降低错配概率,同时引入umi标记原始dna模版。
41.在流程初始阶段,对cfdna进行末端修复,并采用连接的方式而非通过pcr扩增的方式,将包括i5端通用引物结合靶点序列、i5端样本标签和特异性标签(umi)序列的接头一并连接到dna片段上,可大幅降低pcr扩增所放大的误差,引入的特异性标签(umi)可以区分不同来源的dna模版,达成精准定量的效果
42.步骤三、第一轮磁珠纯化:使用纯化磁珠对上述连接反应后的产物进行纯化。上述反应结束后,产物中掺杂有残余的酶、buffer、接头、接头二聚体等杂质,会对后续实验流程产生影响,因此需要对产物进行纯化操作。优选地,本实施例采用beckman公司的agencourt ampure xp beads对pcr产物进行纯化。
43.步骤四、第一轮pcr反应:使用特异性探针抑制野生型dna序列的扩增,使用i5端通用引物和i7端预扩增引物,预扩增突变型dna序列。其中,i7端预扩增引物的条数和种类,取决于目标基因上具体检测位点或区域。目标区域越大或待测位点越多,则特异性引物的条数也随之增加。
44.在第一轮pcr反应中,在合适的退火温度条件下,探针先于预扩增引物与dna模版结合,i5端通用引物可与待测序列一条链进行结合,i7端预扩增引物可与待测序列的另一条链结合。对于突变型dna模版序列,在合适的延伸温度条件下,与靶序列结合的i5端通用引物不受探针影响,具有延伸能力;对于野生型序列,在设定的延伸温度条件下,与dna模版靶序列结合的i5端通用引物,其延伸能力被抑制。上述反应过程将突变模版dna进行富集和放大,从而提高了检测的灵敏度。
45.其中,本实施例提供的文库结构和反应体系支持多重pcr反应,根据不同检测位点设计的i7端预扩增引物之间互不影响。
46.步骤五、第二轮磁珠纯化:使用纯化磁珠对上述第一轮pcr反应产物进行纯化。上述pcr预扩增结束后,产物中掺杂有残余的酶、buffer、dntps、镁离子、引物、引物二聚体等杂质,因此需要对产物进行纯化操作。优选地,本实施例采用beckman公司的agencourt ampure xp beads对第一轮pcr反应产物进行纯化。
47.步骤六、第二轮pcr反应:使用i5端通用引物、i7端特异性引物和i7端接头,进一步富集第一轮pcr反应产物,并添加i7端样本标签用以更好地区分不同样本来源,同时添加一段与p7端测序芯片寡核苷酸簇互补的序列。
48.步骤七、第三轮磁珠纯化:使用纯化磁珠对上述第二轮pcr反应产物进行纯化。上述pcr富集结束后,产物中掺杂有残余的酶、buffer、dntps、镁离子、引物、引物二聚体等杂质,因此需要对产物进行纯化操作。优选地,本实施例采用beckman公司的agencourt ampure xp beads对第二轮pcr反应产物进行纯化。
49.步骤八、文库质控和测序:纯化后的文库使用qubit进行定量,同时使用毛细管电泳(如agilent 2100和qsep100)进行文库片段分布质控。质控合格的文库可在相关二代测序仪上进行测序。
50.参见图3,本实施例方法构建所得文库片段示意图如下:文库片段分布主要与i5端通用引物和i7端特异性引物之间的相互位置有关,大致为240bp-370bp之间。
51.实施例2
52.本实施例以外周血循环肿瘤细胞样本dna突变检测为例具体公开了具体的检测方法
53.1)末端修复
54.此部分分为2个步骤,首先在dna聚合酶的作用下对cfdna进行末端修复,使双链dna形成平末端,再分别在5’端进行磷酸化和3’端加da尾。
55.准备如下表的反应体系:
56.组分体积cfdnaxμl无核酸酶水(40-x)μl片段和末端修复缓冲液10μl片段和末端修复酶10μl总体积60μl
57.配制完成后,充分混匀并离心,使用普通pcr仪,反应程序见下表:
58.温度时间20℃30分钟65℃30分钟4℃hold
59.反应结束后,立刻进行接头连接反应。
60.2)接头连接:通过a-t连接的方式,将i5端接头序列连接至模版dna的5’端,用于添加i5端通用引物结合靶点、i5端样本标签、i5端测序引物重叠序列、特异性随机标签(umi)序列和间隔序列。
61.在本实施例中,所用测序平台为illumina平台,i5端通用引物结合靶点序列为测
序芯片p5端寡核苷酸簇互补序列的一部分。
62.其中,i5端接头的序列和结构为:
[0063]5’‑
aatgatacggcgaccaccgagatctacacnnnnnnnnacactctttccctacacgacgctcttccgatctnnnnnnnnnnnnzzzzzzt-3’。
[0064]
其中,n表示i5端样本标签,n表示特异性随机标签序列,z表示间隔序列。
[0065]
准备如下表的反应体系:
[0066]
组分体积上步反应产物60μli5端接头混合液(15μm)5μl无核酸酶水5μl连接缓冲液30μl连接酶10μl总体积110μl
[0067]
配制完成后,充分混匀并离心,使用普通pcr仪,反应程序见下表:
[0068]
温度时间38℃15分钟4℃hold
[0069]
3)第一轮磁珠纯化:反应结束后,使用ampure xp beads对上述连接反应产物进行纯化,使用20μl纯化水进行洗脱。
[0070]
4)第一轮pcr反应:使用特异性探针抑制野生型dna序列的扩增,使用i5端通用引物和i7端预扩增引物预扩增突变型dna序列。
[0071]
准备如下表的反应体系:
[0072]
组分体积上步纯化产物20μl第一轮pcr扩增反应液20μl特异性探针5μli5端通用引物2.5μli7端预扩增引物2.5μl总体积50μl
[0073]
其中,i5端通用引物序列为:aatgatacggcgaccacc;i7端预扩增引物序列根据检测位点进行特异性的设计;特异性探针根据野生型dna序列进行设计。
[0074]
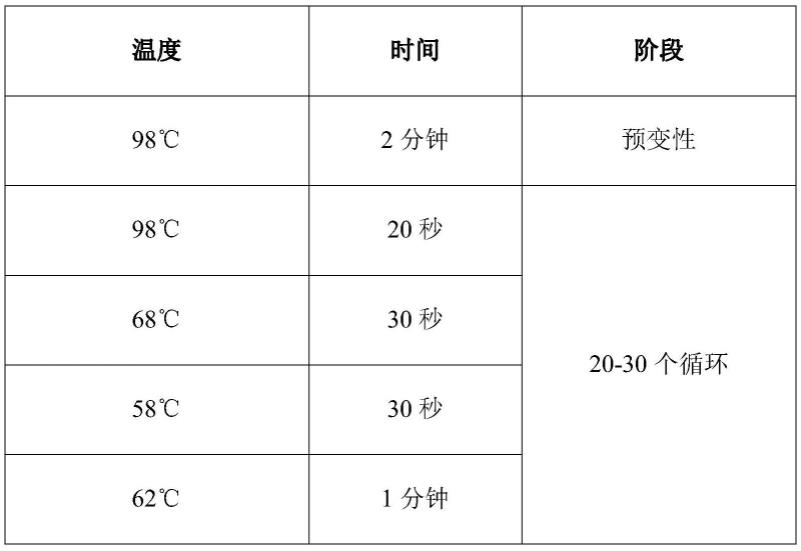
配制完成后,充分混匀并离心,使用普通pcr仪,反应程序见下表:
[0075][0076][0077]
5)第二轮磁珠纯化:第一轮pcr反应结束后,使用ampure xp beads对上述连接反应产物进行纯化,最终使用20μl纯化水进行洗脱。
[0078]
6)第二轮pcr反应:使用i5端通用引物、i7端特异性引物、i7端接头进一步富集第一轮pcr反应产物,旨在进一步提高扩增效率、增加特异性。同时添加i7端样本标签用以更好地区分不同样本来源,同时添加一段与p7端测序芯片寡核苷酸簇互补的序列。
[0079]
准备如下表的反应体系:
[0080]
组分体积上步纯化产物20μl第二轮pcr扩增反应液22.5μli5端通用引物2.5μli7端特异性引物2.5μli7端接头2.5μl总体积50μl
[0081]
其中,i5端通用引物序列为:aatgatacggcgaccacc;i7端特异性引物序列根据检测位点进行特异性的设计,与i7端预扩增引物相比,其碱基位置更靠近检测位点;i7端接头序列为:5
’‑
caagcagaagacggcatacgagatnnnnnnnngtgactggagttcagacgtgnnnn
…
nn-3’。
[0082]
其中,n表示i7端样本标签;n表示与i7端特异性引物部分互补的碱基。
[0083]
配制完成后,充分混匀并离心,使用普通pcr仪,反应程序见下表:
[0084][0085]
7)第三轮磁珠纯化:第二轮pcr反应结束后,使用ampurexp beads对上述连接反应产物进行纯化,最终使用20μl纯化水进行洗脱,即为最终文库。
[0086]
8)文库质控和测序:对最终文库浓度和文库片段分布进行质控,质控合格的文库可在illumina测序仪上进行测序。
[0087]
测序质控结果见下表:
[0088][0089][0090]
所有位点均为100%检出,突变检出情况见下表:
[0091]
[0092][0093]
上表中,chr:突变位点染色体位置;
[0094]
start:突变位点起始位置;
[0095]
end:突变位点终止位置;
[0096]
ref:参考基因组上的碱基序列;
[0097]
alt:检测到的碱基序列;
[0098]
gene:突变位点所在基因;
[0099]
type:基因突变形式;
[0100]
transcript:转录本编号;
[0101]
chgvs:核苷酸突变信息;
[0102]
phgvs:氨基酸突变信息;
[0103]
vaf(%):位点突变频率。
[0104]
对于本领域技术人员而言,显然本发明不限于上述示范性实施例的细节,而且在不背离本发明的精神或基本特征的情况下,能够以其他的具体形式实现本发明。因此,无论从哪一点来看,均应将实施例看作是示范性的,而且是非限制性的,本发明的范围由所附权利要求而不是上述说明限定,因此旨在将落在权利要求的等同要件的含义和范围内的所有变化囊括在本发明内。不应将权利要求中的任何附图标记视为限制所涉及的权利要求。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1