一种阳离子型共聚氨基酸及其制备方法
1.本发明涉及生物医用高分子材料领域,尤其涉及一种阳离子型共聚氨基酸及其制备方法。
背景技术:
2.阳离子型高分子作为一类具有表观正电荷的高分子,与负电性的生物大分子具有强静电相互作用,在基因转染、组织工程等生物领域具有广泛的应用前景,尤其在组织工程领域,传统的组织工程支架缺乏生物组织粘性,容易脱落。由于细胞、组织表面均表现负电性,因此,阳离子型高分子可以通过静电相互作用与生物组织产生粘性。
3.目前的商品化的阳离子型高分子主要包括合成高分子中的聚(甲基)丙烯酸酯类、聚氨基酸类以及天然高分子中的壳聚糖,其中,聚氨基酸类材料具有良好的生物可降解性能,降解产物为氨基酸不会产生无菌性炎症,且能够有效避免天然高分子存在的免疫原性风险,在阻止工程应用中具有广泛应用前景。然而,阳离子聚合物存在两大缺点,一是具有细胞毒性,其来源于高电荷密度对细胞造成的渗透压冲击及对细胞膜的破坏性,二是降解速率难以调控,大多数阳离子聚合物均不可生物降解,少数主链可被酶降解的聚合物无法有效调控其酶降解速率。
4.现有技术中,尽管可以通过对阳离子聚合物中的正电性基团进行化学修饰降低正电荷密度,从而降低细胞毒性,但是往往难以兼顾生物相容性及材料理化性能,而且提高了制备合成上的复杂性,另一方面对酶降解速率的控制仍存在问题,限制了阳离子聚合物在生物医用领域中的应用。
5.因此,现有技术仍有待于改进和发展。
技术实现要素:
6.鉴于上述现有技术的不足,本技术的目的在于提供一种阳离子型共聚氨基酸及其制备方法、应用,旨在解决现有技术中所制备的阳离子聚合物对细胞毒性强,制备复杂,缺乏对酶降解速率的控制,限制了阳离子聚合物在生物医用领域的应用的技术问题。
7.本发明解决技术问题所采用的技术方案如下:
8.第一方面,本技术提供一种阳离子型共聚氨基酸,其中,所述阳离子型共聚氨基酸的结构为:
9.其中,n为5~500,m为5~500,r为氢原子、烷基、取代烷基或烷氨基中的一种,r'为烷基或取代烷基中的一种,且r与r'为不同的取代基团。
10.在上述实现方式中,通过亲核试剂引发氨基酸-nca和n-取代甘氨酸-nca进行开环聚合,得到阳离子型共聚氨基酸,引入电中性的n-取代甘氨酸的聚合物长链分子,能够有效稀释氨基酸的聚合物长链分子的电荷密度,同时,通过对所述阳离子型共聚氨基酸主链上
的酰胺键上的n原子的取代基进行设计,或是通过对共聚氨基酸中氨基酸单元数n以及n-取代甘氨酸的单元数m进行调控,可实现对所制备的所述阳离子型共聚氨基酸的细胞毒性及酶降解速率的调控。
11.可选地,所述取代烷基的取代基为c1~c
12
的烷基、c1~c
12
的烷氧基、c1~c
12
的烯基或卤素中的至少一种。
12.可选地,所述阳离子型共聚氨基酸的分子量分布为1.01~4.0。
13.第二方面,基于同样的发明构思,本技术还提供一种阳离子型共聚氨基酸的制备方法,其中,包括步骤:
14.通过亲核试剂引发氨基酸-nca和n-取代甘氨酸-nca进行开环聚合,制备得到阳离子型共聚氨基酸。
15.在本实现方式中,首先,通过现有技术中制备氨基酸n-羧基环内酸酐(即氨基酸-nca)的方法,制备得到相应的氨基酸-nca和n-取代甘氨酸-nca,其后,通过亲核试剂引发氨基酸-nca和n-取代甘氨酸-nca进行开环聚合,得到阳离子型共聚氨基酸,制备方法简单,易于实现。
16.可选地,所述通过亲核试剂引发氨基酸-nca和n-取代甘氨酸-nca进行开环聚合,制备得到阳离子型共聚氨基酸的步骤还包括,对所述阳离子型共聚氨基酸上带有保护基团的伯胺基进行保护基团的脱除或对所述阳离子共聚氨基酸上的烯基进行后修饰。
17.在上述实现方式中,通过对所述阳离子型共聚氨基酸上带有保护基团的伯胺基进行保护基团的脱除或对所述阳离子共聚氨基酸上的烯基进行后修饰,使得所述阳离子型共聚氨基酸上的化学基团更多样化,有利于后续对所述阳离子型共聚氨基酸的应用。
18.可选地,所述保护基团为叔丁氧羰基、三氟乙酰基、苄氧羰基、芴甲氧羰基中的至少一种。
19.可选地,所述后修饰为通过巯烯点击反应使烯基与巯基发生反应。
20.可选地,所述亲核试剂为小分子胺类、含胺基的高分子或硅烷基金属盐中的至少一种。
21.第三方面,基于同样的发明构思,本技术还提供一种水凝胶制备方法,其中,采用所制备的阳离子型共聚氨基酸以化学或物理交联剂进行交联得到。
附图说明
22.图1为发明中实施例1中所制备的阳离子共聚氨基酸的结构示意图。
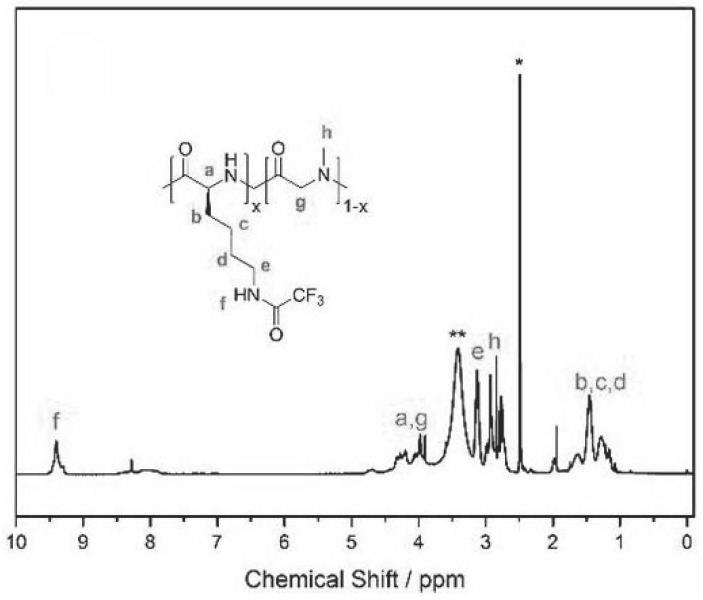
23.图2为发明中实施例1中所制备的阳离子共聚氨基酸的核磁共振氢谱图。
24.图3为本发明实施例2中所制备的阳离子共聚氨基酸的cck-8实验图。
25.图4为本发明实施例2中所制备的阳离子共聚氨基酸的酶降解曲线图。
26.图5为本发明实施例9中所制备的基于阳离子共聚氨基酸水凝胶的活死细胞染色图。
具体实施方式
27.为了便于理解本技术,下面将参照相关附图对本技术进行更全面的描述。附图中给出了本技术的较佳实施方式。但是,本技术可以以许多不同的形式来实现,并不限于本文
所描述的实施方式。相反地,提供这些实施方式的目的是使对本技术的公开内容理解的更加透彻全面。
28.除非另有定义,本文所使用的所有的技术和科学术语与属于本技术的技术领域的技术人员通常理解的含义相同。本文中在本技术的说明书中所使用的术语只是为了描述具体的实施方式的目的,不是旨在于限制本技术。
29.基于现有技术中,阳离子聚合物对细胞毒性与酶降解速率缺乏控制,大大限制了阳离子聚合物在生物医用领域中的应用的问题,本发明提供了一种阳离子型共聚氨基酸及其制备方法、应用,如图1所示,所述阳离子型共聚氨基酸的结构为:其中,n为5~500,m为5~500,r为氢原子、烷基、取代烷基或烷氨基中的一种,r'为烷基或取代烷基中的一种,且r与r'为不同的取代基团。
30.本实施例中,当r为烷氨基时,r'优选为烷基或取代烷基中的一种,当r为氢原子、烷基、取代烷基中的一种时,r'优选为胺基烷硫基取代烷基,r与r'为不同的取代基团。本发明中通过引入电中性的n-取代甘氨酸的聚氨基酸共聚物,能够有效稀释阳离子型聚氨基酸的电荷密度,有效降低细胞毒性,通过对共聚氨基酸中氨基酸单元数n以及n-取代甘氨酸的单元数m进行调控,可实现对所述阳离子型共聚氨基酸的细胞毒性及酶降解速率的同时调控。
31.在一些实施方式中,所述阳离子型共聚氨基酸的分子量分布为1.01~4.0,所述取代烷基的取代基为c1~c
12
的烷基、c1~c
12
的烷氧基、c1~c
12
的烯基或卤素中的至少一种。
32.进一步地,还提供有一种阳离子型共聚氨基酸的制备方法,其中,包括步骤:
33.(1)通过已报道方法(a.j polym sciapolym chem,2012,50,3743
–
3749b.biomacromolecules 2018,19,2109-2116)制备氨基酸-nca和n-取代甘氨酸-nca;
34.(2)通过亲核试剂引发氨基酸-nca和n-取代甘氨酸-nca进行开环聚合,制备得到阳离子型共聚氨基酸。
35.本发明中,首先,通过现有技术中的制备氨基酸n-羧基环内酸酐(即氨基酸-nca)的方法,制备得到相应的氨基酸-nca和n-取代甘氨酸-nca,所述l-氨基酸-nca的结构式为所述n-取代甘氨酸-nca的结构式为其中,n为5~500,m为5~500,r为氢原子、烷基、取代烷基或烷氨基中的一种,r'为烷基或取代烷基中的一种,r与r'为不同的取代基团,当r为烷氨基时,r'优选为烷基或取代烷基中的一种,当r为氢原子、烷基、取代烷基中的一种时,r'优选为胺基烷硫基取代烷基,r与r'为不同的取代基团。
36.其后,通过亲核试剂引发氨基酸-nca和n-取代甘氨酸-nca进行开环聚合,得到阳离子型共聚氨基酸,其中,所述亲核试剂为小分子胺类或含胺基的高分子中的至少一种,所述开环聚合的温度为0~110℃,开环聚合的时间为5分钟~5天,反应方程式如下所示:
[0037][0038]
在一些实施方式中,所述通过亲核试剂引发l-氨基酸-nca和n-取代甘氨酸-nca进行开环聚合,得到阳离子型共聚氨基酸的步骤,还包括有对所述阳离子型共聚氨基酸上带有保护基团的伯胺基进行保护基团的脱除或对所述阳离子共聚氨基酸上的烯基进行后修饰。
[0039]
本实施例中,对所述阳离子型共聚氨基酸上带有保护基团的伯胺基进行保护基团的脱除的步骤中,脱除的所述伯胺基团上的保护基团为叔丁氧羰基、三氟乙酰基、苄氧羰基、芴甲氧羰基中的至少一种,对所述阳离子共聚氨基酸上的烯基进行后修饰通过巯烯点击反应实现,巯烯基点击反应是利用自由基引发巯基与烯基间的偶联形成硫醚结构并引入修饰的官能团的反应,其反应方程式如下所示:
[0040][0041]
在一些实施方式中,还提供有一种水凝胶,所述水凝胶采用上述所制备的阳离子型共聚氨基酸通过化学或物理交联制备而成。
[0042]
本实施例中,通过向所述阳离子型共聚氨基酸中加入能够与所述阳离子型共聚氨基酸分子侧链上的胺基形成共价键或物理相互作用的多官能度小分子或高分子,其中多官能度表示官能度为2及以上,从而使得所述阳离子型共聚氨基酸分子链间发生交联形成水凝胶。
[0043]
以下结合具体实施例对本发明进行进一步的说明。
[0044]
各实施例中所制备的聚合物的数均分子量及分子量分布采用gpc(凝胶渗透色谱)进行检测,其中,以n’n-二甲基甲酰胺作为流动相,温度设定为40℃,流速为1.0ml/min。
[0045]
实施例1聚(三氟乙酰-l-赖氨酸-co-肌氨酸)的合成
[0046]
在反应器中加入1.0g(3.7mmol)的三氟乙酰-l-赖氨酸-nca、425mg(3.7mmol)的肌氨酸-nca,16.7mg(0.1mmol)lihmds以及20mln’n-二甲基甲酰胺,密封后置于60℃油浴中反应12h,反应后将混合物倒入100ml无水乙醚中沉淀、过滤,其后,进行真空干燥,即得到聚(三氟乙酰-l-赖氨酸-co-肌氨酸),收率为80%。采用gpc测得所制备的聚(三氟乙酰-l-赖氨酸-co-肌氨酸)的数均分子量为10000,分子量分布为1.2,另外,根据如图2所示的所制备的聚(三氟乙酰-l-赖氨酸-co-肌氨酸)的氢核磁图谱(cdcl3),可证明成功制备得到聚(三氟乙酰-l-赖氨酸-co-肌氨酸),其中,经过计算,三氟乙酰-l-赖氨酸的单元比例为50%,与投料比相吻合。
[0047]
实施例2聚(l-赖氨酸-co-肌氨酸)的合成
[0048]
在反应器中加入0.5g上述实施例1中所制备得到的聚(三氟乙酰-l-赖氨酸-co-肌氨酸)(含1.6mmol胺基),0.3g氢氧化钠及20ml四氢呋喃,密封后置于60℃油浴中反应12h,反应后将混合物倒入100ml无水乙醚中沉淀、过滤,其后,进行真空干燥,即得到聚(l-赖氨酸-co-肌氨酸),收率为85%。采用gpc测得所制备的聚(l-赖氨酸-co-肌氨酸)的数均分子量为8000,分子量分布为1.2.
[0049]
实施例3聚(三氟乙酰-l-赖氨酸-co-叔丁氧基-n-甲基甘氨酸)的合成在反应器中
加入692mg(3.7mmol)的叔丁基-l-丝氨酸-nca、692mg(3.7mmol)的叔丁氧基-n-甲基甘氨酸-nca,16.7mg(0.1mmol)的lihmds及20ml二甲基甲酰胺,密封后置于60℃油浴中反应12h,反应后将混合物倒入100ml无水乙醚中沉淀、过滤,其后,进行真空干燥,即得到聚(三氟乙酰-l-赖氨酸-co-叔丁氧基-n-甲基甘氨酸),收率为70%。采用gpc测得所制备的聚(叔丁基-l-丝氨酸-co-n烯丙基甘氨酸)的数均分子量为12000,分子量分布为1.3。
[0050]
实施例4聚(l-赖氨酸-co-n-羟甲基甘氨酸)的合成
[0051]
在反应器中加入560mg实施例3中所制备的聚(三氟乙酰-l-赖氨酸-co-叔丁氧基-n-甲基甘氨酸)(含1.6mmol胺基),300mg的氢氧化钠以及20ml的四氢呋喃,密封后置于60℃油浴中反应12h,反应后将混合物倒入100ml无水乙醚中沉淀、过滤,其后,进行真空干燥,即得聚(l-赖氨酸-co-n-羟甲基甘氨酸),收率为80%。采用gpc测得所制备的聚(l-赖氨酸-co-n-羟甲基甘氨酸)的数均分子量为10000,分子量分布为1.3。
[0052]
实施例5聚(甘氨酸-co-n-烯丙基甘氨酸)的合成
[0053]
在反应器中加入374mg(3.7mmol)的甘氨酸-nca、522mg(3.7mmol)的n-烯丙基甘氨酸-nca及20mln’n-二甲基甲酰胺,16.7mg(0.1mmol)lihmds,密封后置于60℃油浴中反应12h,反应后将混合物倒入100ml无水乙醚中沉淀、过滤,其后,进行真空干燥,即得到聚(甘氨酸-co-n烯丙基甘氨酸),收率为80%。采用gpc测得所制备的聚(甘氨酸-co-n烯丙基甘氨酸)的数均分子量为11000,分子量分布为1.1。
[0054]
实施例6聚(甘氨酸-co-n-胺基乙硫基丙基甘氨酸)的合成
[0055]
在反应器中加入185mg上述实施例5中制备得到的聚(甘氨酸-co-n-烯丙基甘氨酸)(含1.2mmol乙烯基),5mg(0.02mmol)安息香二甲醚,1g的巯基乙胺盐酸盐以及20ml四氢呋喃,在氮气气氛下,于270nm的紫外灯下反应4h,反应完成后,将混合物溶液置于截留分子量为1000da的透析袋中,在去离子水中透析3天,其后,冻干得到产物。采用gpc测得所制备的聚(甘氨酸-co-n-胺基乙硫基丙基甘氨酸)的数均分子量为14000,分子量分布为1.2。
[0056]
实施例7聚(叔丁基-l-丝氨酸-co-n-烯丙基甘氨酸)的合成
[0057]
在反应器中加入692mg(3.7mmol)的叔丁基-l-丝氨酸
‑‑
nca、522mg(3.7mmol)的n-烯丙基甘氨酸-nca,16.7mg(0.1mmol)lihmds及20mln’n-二甲基甲酰胺,,密封后置于60℃油浴中反应12h,反应后将混合物倒入100ml无水乙醚中沉淀、过滤,其后,进行真空干燥,即得到聚(叔丁基-l-丝氨酸-co-n-烯丙基甘氨酸),收率为70%。采用gpc测得所制备的聚(叔丁基-l-丝氨酸-co-n烯丙基甘氨酸)的数均分子量为15000,分子量分布为1.3。
[0058]
实施例8聚(l-丝氨酸-co-n-胺基乙硫基丙基甘氨酸)的合成
[0059]
在反应器中加入288mg实施例7中所制备的聚(叔丁基-l-丝氨酸-co-n烯丙基甘氨酸)(含1.2mmol乙烯基),溶于10ml三氟乙酸,密封后置于60℃油浴中反应12h,反应后将混合物倒入100ml无水乙醚中沉淀、过滤,得到聚合物,其后,向制备得到的聚合物中加入5mg dmpa,1g巯基乙胺盐酸盐,20ml的四氢呋喃,在氮气气氛下,270nm的紫外灯下反应4h,反应完成后将混合物溶液置于截留分子量为1000da的透析袋中,在去离子水中透析三天,冻干即得聚(l-丝氨酸-co-n-胺基乙硫基丙基甘氨酸)。采用gpc测得所制备的聚(l-丝氨酸-co-n-胺基乙硫基丙基甘氨酸)的数均分子量为16000,分子量分布为1.3。
[0060]
实施例9基于席夫碱交联的阳离子共聚氨基酸水凝胶的制备
[0061]
将200mg实施例2中所制备得到的聚(l-赖氨酸-co-肌氨酸)与200mg的两端基采用
醛基修饰的聚乙二醇(分子量为2000,含0.2mmol醛基)溶解于10ml的去离子水中,在室温下反应24h,得到水凝胶。
[0062]
实施例10基于双键交联的阳离子共聚氨基酸水凝胶的制备
[0063]
将200mg实施例6中所制备得到的聚(甘氨酸-co-n-胺基乙硫基丙基甘氨酸),20mg的二甲氨基吡啶,20mg的1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,3mg的丙烯酸溶解于10ml的四氢呋喃溶剂中,室温反应下反应10h,其后,倒入100ml的无水乙醚溶液中沉淀、过滤,得到聚合物,向所制备的聚合物中加入200mg的两端基采用丙烯酸酯基修饰的聚乙二醇(分子量为2000,含0.2mmol双键)以及10mg的过硫酸钾,溶解于10ml的水中,在70℃下反应24h,制得水凝胶。
[0064]
实施例11基于物理交联的阳离子共聚氨基酸水凝胶的制备
[0065]
将200mg实施例2中所制备得到的聚(l-赖氨酸-co-肌氨酸)与200mg的两端基采用羧基修饰的聚乙二醇(分子量为2000,含0.2mmol羧基)溶解于10ml的去离子水中,调节ph值并在室温下放置24h,得到水凝胶。
[0066]
进一步地,对实施例中所制备的阳离子共聚氨基酸、基于阳离子共聚氨基酸制备的水凝胶进行表征,其中,图2为实施例2中所制备的阳离子共聚氨基酸的核磁氢谱;图3为实施例2中所制备的阳离子共聚氨基酸的cck-8实验图,图4为实施例2中所制备的阳离子共聚氨基酸的酶降解曲线图,从图3、4中的数据可看出,所制备的聚(l-赖氨酸-co-肌氨酸)不仅可以调控细胞毒性,且可通过调控聚(l-赖氨酸-co-肌氨酸)中l-赖氨酸的比例还调整所制备的聚(l-赖氨酸-co-肌氨酸)的酶降解速率。图5为实施例9中所制备的基于阳离子共聚氨基酸的水凝胶的活死细胞染色图,图中发光点即代表的是活细胞,表明了所制备的基于阳离子共聚氨基酸水凝胶具有良好的生物相容性,可用于组织工程支架。
[0067]
综上所述,本发明中提供了一种阳离子型共聚氨基酸及其制备方法、应用,通过引入电中性的n-取代甘氨酸的聚合物长链分子,能够有效稀释阳离子型聚氨基酸的电荷密度,同时,通过对所述阳离子型共聚氨基酸主链上的酰胺键上的n原子的取代基进行设计,或是通过对共聚氨基酸中氨基酸单元数n以及n-取代甘氨酸的单元数m进行调控,可实现对所述阳离子型共聚氨基酸的细胞毒性及酶降解速率的同时调控,且基于所述阳离子型共聚氨基酸所制备的水凝胶的生物相容性好,可进行推广使用。
[0068]
应当理解的是,本发明的应用不限于上述的举例,对本领域普通技术人员来说,可以根据上述说明加以改进或变换,所有这些改进和变换都应属于本发明所附权利要求的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1