一种基于重组酶聚合酶扩增与CRISPR/Cas12a耦合的DNA甲基化检测方法
一种基于重组酶聚合酶扩增与crispr/cas12a耦合的dna甲基化检测方法
技术领域
1.本发明涉及基因检测技术领域,具体涉及一种基于重组酶聚合酶扩增与 crispr/cas12a耦合的dna甲基化检测方法。
背景技术:
2.表观遗传学的研究已经深入到各个领域,在疾病机制,诊断治疗,药物研发、动植物性状改良等方面均获得了令人瞩目的成就和显著的突破。dna甲基化是最具特征和最常见的表观遗传修饰,主要是基因组dna上的胞嘧啶第5位碳原子和甲基间的共价结合,胞嘧啶由此被修饰为5甲基胞嘧啶,dna甲基化与基因表达和基因稳定性有关。不同于基因突变,dna甲基化修饰是一种可逆的修饰,这使得它成为疾病治疗时重要的潜在靶点。目前,对dna甲基化研究的进展表明,许多编码药物代谢酶、药物运载体、核受体及药物靶点的基因受到dna 甲基化影响,而以dna甲基化为靶向的药物研发也越来越受到关注,临床试验结果也显示dna甲基化药物治疗可减少不良反应以及稳定药效,因此,对dna 甲基化的检测研究有利于新药开发以及发现更多药物靶点。
3.目前,已经开发了用于分析dna甲基化的多种分子检测方法,包括甲基化敏感限制性内切酶(msre)-聚合酶链反应(pcr)、亚硫酸氢盐测序和质谱法等。这些方法主要基于亚硫酸氢盐转化或甲基化敏感限制性内切酶(msre)消化原理。亚硫酸氢盐转化反应被认为是测定dna甲基化的金标准方法,主要是通过亚硫酸氢盐将未甲基化的胞密啶转化为脲嘧啶。但是该过程需要在酸性和高温条件下进行,这会导致dna降解和检测失败。而基于msre原理的技术提供了一种不依赖于亚硫酸氢盐来测定dna甲基化的方法。msre是一类限制性内切酶,其识别位点对甲基化碱基敏感,因此可以区分甲基化和非甲基化序列,并且该反应是在温和的条件下进行,避免了dna降解。然而,来自癌细胞的甲基化dna 含量很低,甚至会低至0.1%,大量的dna未甲基化。因此,基于msre消解的dna甲基化检测的灵敏度和准确性有待进一步提高。
技术实现要素:
4.针对现有技术存在的上述不足,本发明的目的在于提供一种基于重组酶聚合酶扩增与crispr/cas12a耦合的dna甲基化检测方法,以解决现有技术对dna 甲基化检测容易导致dna降解和检测失败、对低浓度的甲基化dna检测灵敏度和准确性不够的问题。
5.为了解决上述技术问题,本发明采用如下技术方案:
6.一种基于重组酶聚合酶扩增与crispr/cas12a耦合的dna甲基化检测方法,其特征在于,包括如下步骤:
7.(1)转录合成crrna:将2μl、10μm的t7启动子、2μl、10μm的crrna 模板和16μl无酶水配制成混合物,放在95℃下退火5min,缓慢冷却至室温;加入10μl、10mm的核苷三磷酸和1.5μl的t7 rna聚合酶,在37℃下转录反应16小时;然后在上述反应液中加入2μl的脱氧核
糖核酸酶i,37℃孵育2小时,对crrna模板进行降解;降解后,用mirna纯化试剂盒纯化产物,并对其进行定量,得到crrna;
8.(2)对dna靶基因和质粒进行甲基化处理:dna靶基因和质粒都是双链,将2μl、32mm的s-腺苷甲硫氨酸、5μl的neb生物缓冲液2、2μl、100u 的cpg甲基转移酶,100nm dna靶基因或2μg目标质粒和无酶水混合成50μl 反应液,然后在37℃孵育5小时;加入2μl、100u的cpg甲基转移酶和1μl、 32mm的s-腺苷甲硫氨酸,37℃继续孵育12小时,然后在65℃反应20分钟后终止反应,得到处理后的甲基化dna或甲基化质粒,将甲基化dna作为电泳验证靶标,甲基化质粒作为检测的目标dna;
9.(3)核酸内切酶裂解目标dna:将2μl步骤(2)处理后的目标dna、1μl 的neb限制性内切酶缓冲液、10u的hhai酶混合,在37℃反应10~70分钟,然后加热至65℃孵育25分钟终止消化,得到消化产物;
10.(4)rpa和单链dna产物的生成:用rpa试剂盒对步骤(3)的消化产物进行扩增,得到双链dna扩增产物,然后在lambda核酸内切酶处理下得到 ssdna;
11.(5)crispr/cas12a检测:将9μl无酶水,2μl cas12酶反应缓冲液,2 μl、1μm的步骤(1)得到的crrna,2μl、1μm的cas12a,1μl、10μm的荧光探针和4μl步骤(4)得到的扩增产物混合,37℃孵育10~50分钟,加入 80μl的双蒸水后对得到的物质进行荧光检测分析,得到目标dna的检测结果。
12.与现有技术相比,本发明具有如下有益效果:
13.1、本发明提供了一种基于核酸内切酶辅助的重组酶聚合酶扩增与 crispr/cas12a(e-pfrpa/cas)耦合无pam位点dna甲基化检测新方法,与传统的甲基化特异性pcr相比,基于核酸内切酶hhai的rpa反应条件温和,检测速度快,在dna甲基化检测中表现出良好的性能,能够对低浓度的甲基化水平进行准确检测,具有很好的应用前景。
14.2、本发明所述方法中利用hhai的特异性,能够对甲基化和非甲基化dna 进行有效区分,同时,rpa结合lambda酶的高扩增能力可以产生大量的ssdna,然后获得的ssdna可以被cas12a识别,识别过程中不需要pam位点,从而避免了crispr/cas12a对pam位点的依赖;并且crispr/cas12a的高特异性和自放大能力进一步提高了传感性能,从而使本发明所述检测方法具有较高的灵敏度和选择性,良好的抗干扰能力和重复性。
附图说明
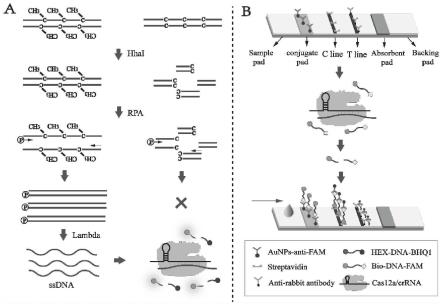
15.图1a为本发明e-pfrpa/cas检测dna甲基化策略示意图;图1b为其在横向流动分析中的应用图。
16.图2a为hhai消化的page图像,泳道1:raffs1a-mc/hhai,泳道 2:raffs1a-c/hhai;图2b为rpa和lambda外切酶处理的page图像,泳道 3:raffs1a-mc/hhai/rpa;泳道4:raffs1a-c/hhai/rpa;泳道5: raffs1a-c/hhai/rpa/lambda;泳道6:raffs1a-mc/hhai/rpa/lambda;图 2c为raffs1a序列中hhai的识别位点图。
17.图3a为不同样品基于e-pfrpa/cas体系的荧光光谱;图3b为不同样本基于e-pfrpa/cas系统的lfa检测图像。
18.图4为对本发明所述方法进行优化后的结果图:图4a为crispr/cas12a的裂解时间、图4b为hhai的消化时间、图4c为rpa的扩增时间、图4d为lambda 的浓度。
19.图5a为目标raffs1a-mc在0fm~20pm不同浓度下的荧光光谱;图5b 为2fm~20pm不同浓度raffs1a-mc的荧光响应,其中插图为2fm~20pm 范围内荧光强度(δf)与raffs1a-mc浓度的对数之间的线性关系;误差条表示三个独立测量值的标准差。
20.图6a为甲基化水平在0~100%范围内的荧光光谱;图6b为输入甲基化水平与测量甲基化水平之间的线性拟合曲线;图6c为浓度为2pm时不同目标 (rassf1a-mc、rassf1a-c、随机dna)的荧光光谱;图6d为2pm浓度下不同靶点(rassf1a-mc、rassf1a-c、随机dna)的荧光反应;误差条表示三个独立测量值的标准差.
21.图7a为2pm浓度下6个实验组的荧光响应,图7b为其对应的荧光值,误差条表示三个独立测量值的标准差.
22.图8a为检测甲基化dna和未甲基化dna的侧流法示意图;图8b为20 fm~20pm不同浓度rassf1a-mc下的lfa条带结果;图8c为不同dna靶点的lfa条带结果。
23.图9a为浓度为200ng/μl时mcf-7癌细胞和正常lo2细胞基因组dna的荧光光谱;图9b为浓度为200ng/μl时,mcf-7癌细胞和正常lo2细胞基因组 dna的荧光响应值,以及显示了浓度为500ng/μl的mcf-7和lo2细胞基因组 dna的lfa条带结果;误差条表示三个独立测量值的标准差。
具体实施方式
24.下面将结合附图和实施例对本发明作进一步说明。
25.一、一种基于重组酶聚合酶扩增与crispr/cas12a耦合的dna甲基化检测方法
26.本发明通过研究发现,现有用于分析dna甲基化的多种分子检测方法在实际使用中存在不少缺点。为了解决现有检测方法中的缺点,本发明考虑首先来自癌细胞的甲基化dna含量很低,需要对其进行扩增。核酸扩增是一种很有前途的目标信号扩增策略。在众多的扩增技术中,滚环扩增(rca)、指数扩增反应(expar)和重组酶聚合酶扩增(rpa)等等温扩增技术都能对目标信号进行有效扩增,与传统的聚合酶链反应(pcr)检测需要热循环过程相比,等温扩增可以在等温条件下进行,并且扩增效率高。但rca所需的锁式探针成本较高,背景信号容易降低检测的灵敏度;而指数扩增反应的特异性和灵敏度不足,限制了其在实际临床检测中的进一步推广和应用。为此,本发明采用rpa进行等温扩增,但仅仅如此还不够。本发明考虑将rpa与其他工具相结合,以进一步提高其准确性、灵敏度和选择性。为此,考虑采用clustered regularly interspaced short palinmicrepeats成簇规律间隔短回文重复序列和其相应的cas蛋白(crispr)/cas系统,将核酸扩增与crispr/cas系统相结合,以求进一步提高检测的灵敏度和特异性。但是,目前,基于不同的crispr/cas系统开发了多种技术,如sherlock (cas13)、detectr(cas12或cas14)和holmes(cas12),例如,在v-a型crispr 系统中,crispr/cas12a(cpf1)表现出对双链dna(double-stranded dna, dsdna)的特异性识别能力,并能激发其反式切割活性以检测目标。然而,这些技术针对双链的识别通常需要tttv原间隔子相邻基序(pam)序列来定位目标区域,但是单链的识别却不需要。所以本发明采用基于核酸内切酶辅助的重组酶聚合酶扩增结合crispr/cas12a开发无pam位点(e-pfrpa/cas)的新方法来敏感地测定dna甲基化。在本发明中,对于不同的癌症,在与其相关基因的启动子区域中往往会发生的甲基化。以乳腺癌为例,rassf1a启动子区域的甲基化被认为是乳腺癌的有效生物标志物;同样对于不同的癌症,选择的启动子会
有所不同。为了检测rassf1a的甲基化,甲基化敏感限制性内切酶用于消化含有限制性内切位点的未甲基化dna。当识别位点甲基化时,目标dna不会被切割,结构保持完整,从而触发rpa反应,在数分钟内获得大量产物。同时,为了实现crispr/cas12a的无pam识别,对rpa的前引物进行了5'磷酸盐修饰,因为lambda外切酶能够识别并降解含有5'-磷酸的序列,从而释放出大量ssdna,使获得的ssdna在不含pam位点的情况下被crispr/cas12a识别,激活其反式切割活性,用于荧光检测和横向流动试纸条。e-pfrpa/cas方法具有较高的灵敏度和特异性,可用于实际样品的检测。
27.所以,本发明所述方法具体如下:
28.(1)转录合成crrna:将2μlt7启动子(10μm)、2μlcrrna模板(10μm)和16μl无酶水配制成混合物,放在95℃下退火5min,缓慢冷却至室温;加入10μl的核苷三磷酸(10mm)和1.5μl的t7rna聚合酶,在37℃下转录反应16小时;然后在上述反应液中加入2μl的脱氧核糖核酸酶i,37℃孵育2小时,对crrna模板进行降解;降解后,用mirna纯化试剂盒纯化产物,并对其进行定量,得到crrna;
29.(2)对dna靶基因和质粒进行甲基化处理:dna靶基因和质粒都是双链,将2μl的s-腺苷甲硫氨酸(32mm)、5μl的neb生物缓冲液2、2μlcpg甲基转移酶(100u),100nmdna靶基因(或2μg目标质粒)和无酶水混合成50μl反应液,然后在37℃孵育5小时;加入2μlcpg甲基转移酶(100u)和1μls-腺苷甲硫氨酸(32mm),37℃继续孵育12小时,然后在65℃反应20分钟后终止反应,得到处理后的甲基化dna靶基因(或甲基化质粒),将甲基化dna靶基因作为电泳验证靶标,甲基化质粒作为检测的目标dna;
30.(3)核酸内切酶裂解目标dna:将2μl步骤(2)处理后的目标dna、1μl的neb限制性内切酶缓冲液、10u的hhai酶混合,在37℃反应10~70分钟,然后加热至65℃孵育25分钟终止消化,得到消化产物;
31.(4)rpa和单链dna(ssdna)产物的生成:用rpa试剂盒对步骤(3)的消化产物进行扩增,得到双链dna(dsdna)扩增产物,其中,扩增步骤为:在rpa冻干粉中加入29.5μl的rpa反应缓冲液,制备得到rpa反应液;然后取4μl步骤(3)得到的消化产物,向其中加入5.9μlrpa反应液,0.5μl的反向引物(10μm),0.5μl的正向引物(10μm)和1μl醋酸镁(280mm),在42℃条件下反应15~35分钟,得到反应产物;然后将2μl、0.5u/μl~4u/μl的lambda核酸内切酶和2μl的lambda缓冲液加入反应产物中,在37℃条件下反应20分钟,得到产物ssdna。
32.(5)crispr/cas12a检测:将9μl无酶水,2μlcas12酶反应缓冲液,2μl的步骤(1)得到的crrna(1μm),2μl的cas12a(1μm),1μl的荧光探针(10μm)和4μl步骤(4)得到的扩增产物混合,37℃孵育10~50分钟,加入80μl的双蒸水后对得到的物质进行荧光检测分析,得到目标dna的检测结果。
33.在具体实施时,在步骤(5)中,所述荧光探针为hex-dna-bhq1荧光探针,在490nm激发下用荧光光谱仪测定荧光强度,得到对目标dna的检测结果。当对目标dna进行横向流动分析的即时检测时,在步骤(5)中,所述荧光探针为横向流动探针bio-dna-fam,将横向流动试纸条插入步骤(5)得到的物质中2分钟后,得到对目标dna的检测结果。
34.二、实施例
35.本发明以乳腺癌为例,rassf1a启动子区域的甲基化被认为是乳腺癌的有效生物标志物,通过如下方法对其进行检测。
36.1、材料与试剂
37.rpa试剂盒可从twistdx公司获得。hiscribe t7高效rna合成试剂盒、hhai 核酸内切酶、lambda核酸外切酶和cpg甲基转移酶(m.sssi)购自中国北京的newengland biolabs。mirna纯化试剂盒来自北京天根。hybridetect试纸(mghd 1)由milenia biotec gmbh提供。所有dna序列和质粒均由上海生工生物技术有限公司合成。ezup柱动物基因组dna纯化试剂盒、sanprep柱pcr产品纯化试剂盒和page相关试剂(acryl/bis 30%solution(29:1)、核酸染料、tbe缓冲液、te缓冲液、depc处理水、上样缓冲液和dna分子量标准marker均来自上海生工生物技术有限公司。
38.表1本发明实施例使用的dna序列
[0039][0040]
注:raffs1a-c,raffs1a,random 1,random 2and random 3都是双链 dna.“p”是指磷酸基团修饰。
[0041]
2、crrna以及甲基化靶标的制备
[0042]
体外通过t7转录合成crrna。首先,将2μl t7启动子(10μm)、2μl crrna 模板(10μm)和16μl无酶水配制成混合物,放在95℃下退火5min,缓慢冷却至室温;加入10μl的核苷三磷酸(10mm)和1.5μl的t7 rna聚合酶,在37℃下转录反应16小时;然后在上述反应液中加入2μl的脱氧核糖核酸酶i, 37℃孵育2小时,对crrna模板进行降解;降解后,用mirna纯化试
剂盒纯化产物,并用nanodrop 2000c(thermo fisher)定量。对得到的crrna进行凝胶电泳分析。
[0043]
dna靶基因和质粒经过cpg甲基转移酶处理,处理步骤如下:2μl的s-腺苷甲硫氨酸(32mm)、5μl的neb生物缓冲液2、2μl cpg甲基转移酶(100u), 100nm dna靶基因(或2μg目标质粒)和无酶水混合成50μl反应液,然后在 37℃孵育5小时;加入2μl cpg甲基转移酶(100u)和1μl s-腺苷甲硫氨酸(32 mm),37℃继续孵育12小时,然后在65℃反应20分钟后终止反应,得到甲基化dna靶基因和甲基化质粒。将甲基化dna靶基因作为电泳验证靶标,甲基化质粒作为检测的目标dna;
[0044]
3、hhai核酸内切酶裂解靶标dna
[0045]
用hhai酶消化目标dna(raffs1a-c和raffs1a-mc),反应总体积为10 μl。具体步骤如下,加入目标dna 2μl,neb限制性内切酶缓冲液1μl,hhai 10u和无酶水,37℃反应50分钟,然后在65℃加热25分钟终止消化。
[0046]
4、rpa和ssdna的生成
[0047]
用rpa试剂盒对消化产物进行扩增。首先,在rpa冻干粉中加入29.5μl 的反应缓冲液,制备rpa反应液;然后取4μl消化产物,向里加入5.9μl rpa 反应液,0.5μl(反向引物(10μm),0.5μl正向引物(10μm)和1μl醋酸镁(280 mm),42℃反应25分钟。为了获得ssdna,将2μl lambda核酸内切酶(2u/μl) 和2μl lambda核酸内切酶缓冲液加入上述溶液中,37℃反应20分钟。
[0048]
5、电泳分析
[0049]
用非变性10%聚丙烯酰胺凝胶验证了该方法的反应原理。由于质粒样本基因太长,因此合成了125bp的dna靶基因双链作为电泳分析目标。将反应后的样品与6x的上样缓冲液在室温下混合2分钟,然后移至凝胶孔中。电泳在1 xtbe缓冲液中进行,电压为90v,电泳时间50分钟。最后,电泳凝胶在核酸染料中反应15分钟,用jena uvsolo imager采集图像。
[0050]
6、crispr/cas12a检测
[0051]
荧光光谱和侧向流动检测是基于cas12a的反式切割活性,因此cas12a反应过程如下,反应总体积为20μl:9μl无酶水,2μl cas12酶反应缓冲液,2μl 的crrna(1μm),2μl的cas12a(1μm),1μl的荧光探针(10μmhex-dna-bhq1)和4μl反应产物,37℃孵育30分钟,然后加入80μl的双蒸水,在490nm激发下用荧光光谱仪(perkinelmer)测定荧光强度。对于横向流量检测,荧光探针被横向流动探针(10μm bio-dna-fam)取代。其他反应步骤与上述步骤一致。然后将试纸条插入反应溶液中2分钟,最后用相机拍摄结果。
[0052]
7、细胞培养及基因组dna制备
[0053]
选取人乳腺癌细胞(mcf-7)和正常人肝细胞(lo2)作为真实样本。为了制备基因组dna,这些细胞首先在90%rpmi 1640培养基(hyclone,美国)和10%胎牛血清(solarbio,中国)中培养。当细胞密度达到培养瓶的80%时,提取基因组 dna,用ezup柱动物基因组dna纯化试剂盒纯化。然后将获得的基因组dna 作为目标dna,验证该方法的实际应用能力。
[0054]
三、对检测原理以及结果的讨论
[0055]
1、e-pfrpa/cas检测原理
[0056]
本发明所述检测方法的原理见附图1。从附图1a可以看出,甲基化dna 和未甲基化
dna首先经过hhai外切酶处理,该酶可以识别和消化
“‑
gcgc
‑”
位点。因此,未甲基化的dna会被hhai消化。在本发明中,raffs1a基因启动子含有多个-gcgc-的酶切位点。因此,未甲基化的raffs1a完全被hhai 消化。相反,甲基化raffs1a没有影响,结构保持完整。结构完整的甲基化 raffs1a被用来触发rpa扩增反应。在pra系统中,重组酶与引物结合定位双链中的同源序列,并以指数方式扩增靶区。由于正引物的5'端有一个磷酸基修饰,所有扩增的双链中正义链均有5'-磷酸。lambda外切酶可以降解双链的5'
‑ꢀ
磷酸修饰序列。因此,通过lambda处理可以获得大量的单链ssdna。同时,在 lambda处理下可以避免非特异性放大,有效地过滤了检测背景。由于产生的 ssdna与crrna部分互补,从而激活cas12a的反式切割活性,cas12a可以任意切割附近的荧光报告探针(hex-dna-bhq1)来释放荧光信号。
[0057]
此外,本发明所述方法还可以通过改变报告探针(bio-dna-fam),来用于横向流动分析(lfa)的即时检测。如附图1b所示,biotin和fam在报告探针的两端进行修饰,使其能够与aunps-anti-fam结合。由于在对照线区域(c线)中存在链霉亲和素,可以通过生物素与链霉亲和素结合来捕获aunps-anti-fam/ bio-dna-fam,因此,c线上出现了一条红带。当存在甲基化靶点时,它们可以启动一系列反应产生大量ssdna。然后cas12a的反式切割活性被激活,从而剪切bio-dna-fam报告基因。含有aunps的被剪切报告探针可以被测试线(t 线)上的兔抗人血清白蛋白抗体捕获,t线上会出现红色条纹。相反,在未甲基化的靶区,c线上只有一条红带。基于传感器的检测原理,可以成功区分dna 甲基化,并应用于即时护理平台。
[0058]
2、本发明采用e-pfrpa/cas战略的可行性
[0059]
通过电泳、荧光检测和lfa检测验证e-pfrpa/cas策略的可行性。图2a和图2b为电泳图像。甲基化的raffs1a和未甲基化的raffs1a长度为125bp,未甲基化的raffs1a含有hhai的
“‑
gcgc
‑”
位点,与hhai反应后,raffs1a 被消化成不同的片段。从图2a中可以看出,在泳道2中可以观察到被消化的条带,这与图2c的示意图是一致的。由于其中有些被消化的序列太短导致染色不清楚或者跑出电泳板,比如5bp和7bp的条带,在泳道2看不到。最后一个低迁移率的条带是未完全消化的raffs1a,这刚好对应于泳道1的甲基化 raffs1a,它受到甲基修饰保护不会被内切酶消化。这些结果表明甲基化 raffs1a和非甲基化raffs1a已经成功地被hhai特异性识别。图2b为rpa 反应和lambda外切酶处理的电泳结果图。扩增后,在第泳道3出现一条明显的条带,由于单链结合蛋白和重组酶的干扰,扩增条带较宽,另外两个条带分别是正向引物和反向引物。对于未甲基化的raffs1a,由于不完全消化和非特异性扩增,在4巷也有rpa产物。然而,当添加lambda外切酶时,只有甲基化靶标才能生成ssdna(泳道5),而未甲基化的靶标被消化没有ssdna产生。
[0060]
为了进一步验证基于crispr/cas12a的dna甲基化检测,荧光光谱和lfa 结果如图3a和图3b所示。从图3a中,只有甲基化的raffs1a(raffs1a-mc) 才会产生较强的荧光(曲线c),而未甲基化的raffs1a(raffs1a-c)(曲线b)和空白组(曲线a)的荧光可以忽略不计。上述结果表明,raffs1a-mc可以通过5mc 修饰避免hhai酶切,进而启动rpa反应生成大量的dna双链。加入lambda 后,具有5'-磷酸末端的dna序列可以被识别和降解,留下大量ssdna。因此,获得的ssdna可以激活cas12a的反式切割活性,释放荧光信号。相反, raffs1a-c被hhai消化,hhai不能引发以下一系列反应。在lfa检测中,只有具有raffs1a-mc的实验组在测试线上出现了一条明显的带,这是由于 aunps-anti-fam偶联所致。上述结果表明,e-pfrpa/cas方法能够成功检测dna 甲基化,且背景干扰低,灵敏度和准确性高。
[0061]
3、对本发明所述方法进行优化
[0062]
为了提高该方法的灵敏度,对cas12a的裂解时间、hhai的消化时间、rpa 的扩增时间和lambda的浓度等重要参数进行了优化。首先,研究了cas12a的剪切时间。如图4a所示,荧光强度随着cas12a裂解时间的增加而增强,在30 分钟达到峰值,因此接下来的实验cas12a裂解时间为30分钟。hhai与背景信号有关,消化不完全会导致高背景信号。设置50pm高浓度的dna靶标 (raffs1a-mc和raffs1a-c)和高浓度的酶含量(10u)以确保完全消化。如图4b所示,随着消化时间从10分钟增加到70分钟,背景信号减小,hhai处理 50分钟后荧光信号完全淬灭。因此,未甲基化dna的消化需要hhal处理50分钟。扩增时间也是一个重要参数,与灵敏度密切相关。扩增时间为15~35分钟后测量rpa反应的荧光强度,25分钟后出现饱和荧光信号(图4c)。此外,对 lambda酶的浓度也进行了优化,以便于快速检测。如图4d所示,荧光响应呈浓度依赖性,在2u/μl处达到饱和。因此,选择2u/μl的lambda作为最优条件。
[0063]
4、e-pfrpa/cas检测dna甲基化的性能研究
[0064]
为了研究该方法的性能,在最佳条件下测量了该方法对raffs1a-mc检测的浓度范围。如图5a所示,从0fm到20pm,随着raffs1a-mc浓度的增加,荧光响应逐渐增强。荧光强度(δf)与raffs1a-mc浓度的对数在2fm到20pm 之间存在良好的线性关系(图5b插图)。荧光强度(δf)为减去空白组荧光强度后的值。线性拟合方程为δf=241.3logc+623.3(r=0.994),c为raffs1a-mc (pm)浓度。检测限(lod)计算为0.98fm。与现有技术的dna甲基化检测方法相比,本发明所述方法的检出限较低,检测范围较宽(表2)。这些优异的性能可能得益于以下原因:(1)hhai的特异性识别使甲基化和非甲基化dna能够有效区分;(2)rpa结合lambda酶的高扩增能力可以产生大量的ssdna;(3)获得的 ssdna可以被cas12a识别,不需要pam位点;而crispr/cas12a的高特异性和自放大能力进一步提高了传感性能。
[0065]
表2与其他甲基化检测方法比较
[0066][0067]
现有技术表明,哺乳动物基因组中5mc占胞嘧啶的2-7%,大部分基因处于未甲基化状态。因此,在存在大量未甲基化样本的情况下,有必要区分甲基化区域。首先,将非甲基化靶标(raffs1a-c)浓度设置为20pm,然后将不同浓度的甲基化靶标(raffs1a-mc)加入到非甲基化(raffs1a-c)反应体系中。如图6a 所示,随着甲基化水平的增加,荧光信号逐渐从
0%增加到100%,输入甲基化水平与测量甲基化水平之间存在良好的线性关系。线性拟合方程为y=1.1x+0.41 (r=0.999),y为测量甲基化水平,x为输入甲基化水平。值得注意的是,0.05%的甲基化水平(10fm)可被识别,表明本发明采用的e-pfrpa/cas策略具有较高的准确性和敏感性。
[0068]
选择性和可重复性是传感系统的关键。因此,首先以rassf1a-c等随机 dna作为干扰物,研究本发明所述方法对位点特异性甲基化检测的选择性。从图6c和图6d可以看出,随着rassf1a-mc的存在,出现了一个明显的荧光峰。而在相同浓度的rassf1a-mc(2pm)下,相对于rassf1a-c和随机dna,荧光反应较弱。此外,通过测试5个平行实验组和对照组的荧光反应来考察重复性(图7)。各组间荧光值无明显波动。相对标准偏差(rsd)为2.28%。上述结果表明,本发明所述检测方法具有良好的抗干扰能力和重复性。
[0069]
5、横向流动试验(lfa)对dna甲基化检测
[0070]
横向流动试纸条具有直观、便携的特点,因此对于建立肉眼检测平台具有良好的实际应用潜力。因此,本发明将e-pfrpa/cas方法集成到横向流动试验(lfa) 条带中。图8a为侧流法检测甲基化dna和未甲基化dna的原理图。根据条带的检测原理,当存在甲基化目标(rassf1a-mc)时,在c线上和t线上有两条红色条带。而在未甲基化的靶标(rassf1a-c)中,只有一条红带出现。然后研究了基于e-pfrpa/cas法的lfa灵敏度。如图8b所示,在t线上,随着rassf1a-mc 浓度从0fm到20pm的增加,波段颜色呈现由无色向暗渐变的趋势。然而,c 线上的颜色显示了相反的趋势。低至20fm的rassf1a-mc可以被明显识别。此外,从图8c可以看出,只有rassf1a-mc能在测试线上诱导出红色。 rassf1a-c等随机dna仅在控制线上显示红色。这些结果表明,本发明采用的基于e-pfrpa/cas方法的lfa具有较高的灵敏度和选择性,在资源有限的地区具有较好的应用前景。
[0071]
6、真实样本检测
[0072]
为了评估e-pfrpa/cas检测方法的实用性,在目标溶液中加入20倍稀释的人血清,模拟复杂的生物环境。稀释目标浓度分别为20fm、200fm和2pm。稀释靶标采用e-pfrpa/cas法测定。结果如表3所示,回收率为97%~112.4%。 rsd为2.05%~4.66%,适用于复杂样品的检测。此外,有资料表明rassf1a 启动子的甲基化与乳腺癌密切相关,该甲基化区域(50340771位点-50340676位点)可以影响细胞角蛋白19(ck19)mrna的表达,进而导致肿瘤的发生。因此,为了进一步评价本发明所述方法在实际应用中的可靠性,将e-pfrpa/cas检测应用于真实基因组dna样本。研究了基于该方法检测正常细胞(lo2)和乳腺癌细胞(mcf-7)的基因组dna。如图9a和图9b所示,在相同数量的基因组dna(200 ng/μl)下,mcf-7细胞基因组dna的荧光反应显著高于正常细胞(lo2)的基因组dna,并且在相同数量的基因组dna(500ng/μl)下,mcf-7细胞基因组dna 在t线上也显示出了红色条带,正常细胞(lo2)的基因组dna仅在控制线上显示红色。以上结果表明本发明所述方法对细胞dna甲基化的测定具有较高的敏感性。
[0073]
表3用于人血清样品中dna甲基化检测(rassf1a-mc)
[0074]
加入靶标浓度检测计算浓度回收率(%)rsd(%)20fm19.4fm974.66200fm224.7fm112.44.632pm2.18pm1092.05
[0075]
7、结论
[0076]
综上所述,本发明提供了一种基于核酸内切酶辅助的重组酶聚合酶扩增与 crispr/cas12a(e-pfrpa/cas)耦合无pam位点dna甲基化检测新方法。与传统的甲基化特异性pcr相比,基于hhai的rpa反应条件温和,检测速度快。在不含pam位点的情况下触发crispr/cas12a的反式切割活性。结果表明,本发明所述方法在dna甲基化检测中表现出良好的性能,并可与侧流条带相结合实现poc检测。荧光检测限为0.98fm,可区分低至0.05%的甲基化水平。通过侧流法,肉眼可明显识别20fm的甲基化dna。此外,e-pfrpa/cas可检测到癌细胞的基因组dna甲基化。基于crrna的可编程性,e-pfrpa/cas方法可进一步用于建立多个甲基化位点甚至核酸检测的通用平台,具有很好的应用前景。
[0077]
最后需要说明的是,以上实施例仅用以说明本发明的技术方案而非限制技术方案,本领域的普通技术人员应当理解,那些对本发明的技术方案进行修改或者等同替换,而不脱离本技术方案的宗旨和范围,均应涵盖在本发明的权利要求范围当中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1