一种靶向降解CYP1B1的PROTAC化合物及其制备方法和应用
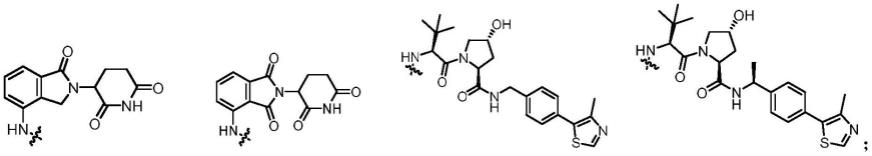
一种靶向降解cyp1b1的protac化合物及其制备方法和应用
技术领域
1.本发明属于药物合成与应用领域,具体涉及一类具有cyp1b1降解活性的小分子-蛋白降解靶向嵌合体及其制备方法以及其在抗肿瘤方面的应用。
背景技术:
2.cyp1b1是细胞色素p450(cyp450)超家族1的一个成员,该家族蛋白还包括cyp1a1和cyp1a2。不同于后两者,cyp1b1通常在肝外组织中,特别是肿瘤组织中过度表达。
3.cyp1b1主要从两个方面介导肿瘤的发生发展。第一方面,cyp1b1会参与代谢tcdd,bap,hahs等,代谢产物多具有dna加合毒性,引起肿瘤发生。第二方面,cyp1b1还参与雌激素的异常代谢。雌二醇通过cyp1a1或cyp1a2转化为2-羟基雌二醇,进而通过儿茶酚-o-甲基转移酶代谢生成2-甲氧基雌二醇,该代谢产物无基因毒性。但是,cyp1b1可以将雌二醇代谢为4-羟基雌二醇,不能被进一步代谢,但是过氧化物酶可以催化其生成雌二醇-3,4-醌,雌二醇-3,4-醌具有dna加成毒性,所以,在雌激素异常代谢引起的癌症机制中,cyp1家族蛋白特别是cyp1b1可能产生了关键作用。此外,还有部分研究证明cyp1b1的过度表达与肿瘤耐药和转移密切相关。因此cyp1b1是一个与肿瘤的发生发现、耐药性、迁移侵袭相关的潜在靶点。
4.肿瘤是威胁人类健康的最大杀手之一,其中,肺癌是发病占比最高的疾病。cyp1b1蛋白在肿瘤细胞系内过表达,以cyp1b1为靶点的抑制剂可对肿瘤细胞的生存,迁移有较强的抑制活性。
5.近年来,人们利用蛋白水解靶向嵌合体(proteolysis targeting chimeras,protac)技术成功实现了对不同蛋白的靶向降解。protac是双功能分子,由三个关键部分组成:
①
靶蛋白(protein of interest,poi)结合配体、
②
e3泛素连酶的招募配体、
③
连接前两者的linker。protac通过招募e3泛素酶来促进靶蛋白的泛素化,随后通过泛素-蛋白酶体途径降解靶蛋白。目前针对protac分子的研发以及优化,主要有下面几个方面:
6.1)靶蛋白配体的选择,靶蛋白的配体主要来源于相应开发的抑制剂,但是往往同一蛋白的抑制剂的种类繁多,需要实验筛选最合适的protac的靶蛋白结合配体。
7.2)e3泛素连接酶的选择,目前已知的人体内的泛素连接酶有600多种,但是目前仅有如mdm2,ciap,crbn,vhl等少数e3连接酶成功应用在protac上,目前针对特定靶蛋白,如何选用e3连接酶,仍然无特定的指导原则,需要筛选和实验尝试才能确定。
8.3)连接linker的优化,这方面包括linker的分子结构,分子量,长度,对于最终protac的降解效果影响是十分巨大的。目前,常用的linker有peg、alkyl、click chemistry linker等等。linker的确定和选择主要基于实验筛选和测试。
9.4)protac分子的溶解性以及透过细胞膜的能力:protac分子由于分子量比传统的抑制剂大很多,往往不具备较好的成药性和药代动力学性质。简化配体的分子量以及在合理范围内优化linker是一种可行的办法。同样需要基于实验筛选和测试。
10.目前还没有进入临床研究阶段的cyp1b1抑制剂报道,也没有进入临床的protac化
合物报道。
技术实现要素:
11.本发明目的在于提供一种靶向降解cyp1b1的protac化合物及其药学上可接受的盐、制备方法和应用。本发明提供的cyp1b1蛋白降解剂具有protac分子结构,能够有效降解cyp1b1蛋白,从而逆转肿瘤细胞的耐药,抑制肿瘤细胞的迁移侵袭。
12.本发明的技术方案如下:
13.本发明的首要目的是提供下述通式i或通式ii所示的靶向降解cyp1b1的protac化合物(cyp1b1-protac化合物)或其药理或生理上可接受的盐。
[0014][0015]
其中:
[0016]
x为-o-或-nhco-;
[0017]
r为卤素、氰基、硝基、酯基、r1、or1、(ch2)nnr1r2、(ch2)nc(o)r1、(ch2)
n c(s)r1、c(o)nr1r2、sr1、s(o)mr1、s(o)2nr1r2、oc(o)r1、oc(o)nr1r2、os(o)2r1、os(o)2nr1r2、nr2c(o)r1、nr2c(o)r1r2、n(r2)s(o)2r1或n(r2)s(o)2nr1r2;
[0018]
r1、r2各自独立地为h、取代的或未取代的烷基、取代的或未取代的c
2-6
烯基、取代的或未取代的c
2-6
炔基、取代的或未取代的c
3-7
环烷基、取代的或未取代的c
5-7
环烯基、取代的或未取代的芳基、取代的或未取代的杂芳基、取代或未取代的3-至15-元杂环基或氨基;其中,各取代是指被选自下组的一个或多个基团所取代:卤素、氰基、硝基、酯基、三氟甲基、三氟乙酰基、三氟甲磺酰基、c
l-6
烷基、c
3-7
环烷基、卤代c
l-6
烷基、c
2-6
烯基、卤代c
2-6
烯基、c
2-6
炔基、卤代c
2-6
炔基、羟基、羟基-c
1-4
烷基、or3、nr3r4、c(o)r3、co(o)r3、c(o)nr3r4、sr3、s(o)mr3、s(o)2nr3r4、oc(o)r3、oc(o)nr3r4、os(o)2r3、os(o)2nr3r4、nr3c(o)r4、nr3c(o)nr4r5、n(r3)s(o)2r4、n(r3)s(o)2nr4r5;
[0019]
r3、r4、r5各自独立地为h、c
1-6
烷基、c
1-6
卤代烷基、c
2-6
烯基、卤代c
2-6
烯基、c
2-6
炔基或卤代c
2-6
炔基;
[0020]
e3连接酶配体是指可以结合e3连接酶的配体分子,e3连接酶包括vhl(von hippel-lindau,希尔佩-林道)和crbn(cereblon基因编码的蛋白)两种,其配体分子包括如下结构:
[0021][0022]
linker为连接基团,表示-亚烷基或-烷氧基,所述-亚烷基或-烷氧基选自以下基团中任一个或它们的任意组合;-(ch2)
n-、-(ch2)
n co-、-nr1(ch2)
n co-、-nr2(ch2)
n-、-(och2ch2o)
n-、-(ch2ch2o)
n-、-(och2ch2och2)
n-、-(ch2ch2och2)
n-、-(ch2ch2och2ch2)
n-、亚烯基、亚炔基、亚环烷基、亚杂芳烃基;其中n表示1至20的自然数,r1、r2各自独立地为h或c
1-10
烷基。
[0023]
进一步的,本发明提供的cyp1b1-protac化合物,其为如下所示化合物或其立体异构体、几何异构体、互变异构体、氮氧化物、水合物、溶剂化物、代谢产物、药学上可接受的盐或前药;
[0024]
[0025]
[0026]
[0027]
[0028][0029]
本发明所述药理或生理上可接受的盐是指,本发明所述的cyp1b1-protac化合物与药理或生理上可接受的酸或碱生成的加成盐。
[0030]
本发明所述卤素包括氟、氯、溴、碘。
[0031]
本发明还提出一种药物组合物,所述的药物组合物包括所述的cyp1b1-protac化合物或其立体异构体、几何异构体、互变异构体、氮氧化物、水合物、溶剂化物、代谢产物、药学上可接受的盐或前药。
[0032]
所述的药物组合物还包括药学上可接受的载体、赋形剂、稀释剂、辅剂、媒介物或其组合。
[0033]
所述药物组合物为注射剂、口服剂、黏膜给药剂。
[0034]
所述的药物组合物进一步包括其它具有治疗或预防肿瘤效果的药物。
[0035]
本发明还提供所述cyp1b1-protac化合物或包含该cyp1b1-protac化合物的药物组合物的应用。具体如下:
[0036]
所述cyp1b1-protac化合物或包含该cyp1b1-protac化合物的药物组合物在制备降解cyp1b1或抑制cyp1b1药物中的应用。
[0037]
所述cyp1b1-protac化合物或包含该cyp1b1-protac化合物的药物组合物在制备治疗或预防cyp1b1相关性疾病药物中的应用。所述cyp1b1相关疾病为肺癌,进一步的,为非小细胞肺癌。
[0038]
所述cyp1b1-protac化合物或包含该cyp1b1-protac化合物的药物组合物在抗肿瘤药物中的应用。所述肿瘤为乳腺癌、白血病、肺癌、肝癌、食管癌、胰腺癌、结直肠癌、胃癌、宫颈癌、脑癌、前列腺癌。进一步的,所述肿瘤为cyp1b1高表达的肿瘤,或耐cyp1b1抑制剂型肿瘤。更进一步的,所述肿瘤为肺癌实体瘤。
[0039]
本发明还提出了通式i或通式ii所示的cyp1b1-protac化合物合成路线,具体包括如下步骤:
[0040]
通式i或通式ii所示的化合物通过pomalidomide或lenanidomide或vhl配体和靶蛋白配体之间通过click反应或酰胺缩合反应或亲核取代反应连接而成,其中,pomalidomide端衍生物的制备方法参考文献chemistry&biology 22,755-763(2015),lenalidomide端的衍生物的制备方法参考j.med.chem(doi:10.1021/acs.jmedchem.6b01816),vhl配体部分的制备方法参考参考文献j.med.chem(doi:10.1021/acs.jmedchem.1c00460)。
[0041]
本发明有益效果:
[0042]
本发明合成并筛选了一种新的化合物,为新型protac降解剂化合物。发明人通过western blot实验证实本发明提供的新型protac降解剂化合物对cyp1b1的降解效果,进一步通过细胞增殖实验及迁移侵袭实验证明该新型protac降解剂化合物有效抑制细胞增殖及迁移侵袭,有潜力成为治疗恶性肿瘤的有效治疗方式。
附图说明
[0043]
图1是本发明化合物p31的1h-nmr光谱图;
[0044]
图2是本发明化合物p31的
13
c-nmr光谱图;
[0045]
图3是本发明实施例化合物的分子在部分细胞系的对cyp1b1的降解效果筛选;
[0046]
图4表示本发明实施例化合物p31的降解cyp1b1作用具有浓度依赖性和时间依赖性;
[0047]
图5表示本发明实施例化合物p31通过泛素-蛋白酶体突进降解cyp1b1蛋白;
[0048]
图6是本发明实施例化合物p31对a549/taxol细胞的体外抑制作用;
[0049]
图7是本发明实施例化合物p31对a549/taxol细胞株的迁移侵袭抑制作用。
具体实施方式
[0050]
下面详细描述本发明的实施例,下面描述实施例是示例性的,旨在用于解释本发明,而不能理解为对本发明的限制。
[0051]
本发明对试验中所用到的材料以及实验方法进行一般性和具体性的描述。虽然为实现本发明的目的所使用的许多材料和操作方法是本领域公知的,但是本发明仍然在此作尽可能的描述。本领域技术人员清楚,在下文中,如未特别说明,所使用的材料和操作方法
是本领域公知的。
[0052]
实施例1
[0053]
靶向降解cyp1b1化合物的合成与结构确认
[0054]
合成路线:
[0055]
[0056][0057]
化合物1的合成
[0058]
将来那度胺(化合物0,0.30g,1.16mmol),戊二酸(0.15g,1.16mmol),2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu,0.66g,1.74mmol),diea(0.3ml,1.74mmol),加入dmf 8ml,室温下搅拌4h,tlc显示反应完全,将反应液倒入水中,冷却静置,有白色固体析出,抽滤,滤饼水洗,乙醚洗两次,干燥,柱色谱分离(dcm:meoh=20:1.v/v),得白色固体化合物1a 100mg,收率:23.1%。
[0059]
化合物1b、1c、1d、1e、1f按相同方法合成。
[0060]
化合物p1的合成
[0061][0062]
将向25ml反应瓶中依次加入中间体1a(100mg,0.27mmol),l1(73mg,0.27mmol),hatu(400mg,0.40mmol),diea(0.07ml,0.40mmol),加入dmf 5ml,室温下搅拌3h,tlc显示反应完全,将反应液倒入水中,乙酸乙酯萃取4次,合并有机层,有机层用饱和碳酸氢钠溶液和饱和食盐水各洗涤3次,无水na2so4干燥,硅胶柱色谱分离,得到白色固体72mg,收率:46.2%。1h nmr(600mhz,dmso-d6)δ11.02(s,1h),10.58(d,j=3.0hz,1h),10.09(s,1h),9.99(s,1h),7.87(dd,j=7.1,3.2hz,4h),7.74(d,j=3.2hz,2h),7.71(d,j=3.2hz,1h),7.56(dd,j=8.1,2.2hz,1h),7.50(d,j=1.8hz,1h),7.34(d,j=8.1hz,2h),7.16(d,j=8.0hz,1h),5.13(dd,j=13.3,5.1hz,1h),4.57
–
4.28(m,2h),2.98
–
2.86(m,2h),2.64
–
2.56(m,1h),2.47(t,j=7.4hz,2h),2.43(t,j=7.3hz,2h),1.94(t,j=7.4hz,2h),1.20(d,j=34.4hz,1h).
13
c nmr(151mhz,dmso-d6)δ173.35,171.67(d,j=31.1hz),168.31,163.90(d,j=20.7hz),160.70,158.87,134.53,134.21(d,j=18.2hz),133.14,130.13,129.13(d,j=5.0hz),128.50,128.37,126.75,125.70,121.04,119.46,118.96,118.79,114.98,99.29,57.84,51.98,46.93,36.07,31.67,27.02,25.44,23.82
–
22.47(m),19.12.hrms(esi,m/z)calcd for c
30h27
n7o5s2[m+na]
+
,652.1515;found,652.1413.
[0063]
化合物p2、p3、p4、p5、p6按相同方法合成。
[0064][0065][0066]
化合物p7、p8、p9、p10的合成方法与p1的合成相同,仅将靶蛋白配体l1变为l2,与相应的1b-d,1f反应得到。
[0067]
[0068]
p7:1h nmr(400mhz,dmso-d6)δ10.99(s,1h),10.43(s,1h),9.89(s,1h),9.77(s,1h),7.90(t,j=2.1hz,1h),7.86(d,j=3.3hz,1h),7.82(dd,j=7.1,2.1hz,1h),7.71(d,j=3.2hz,1h),7.54
–
7.50(m,1h),7.49(d,j=1.8hz,1h),7.47(s,1h),7.25(t,j=8.1hz,1h),7.14
–
7.09(m,1h),5.75(s,1h),5.13(dd,j=13.2,5.1hz,1h),4.45
–
4.28(m,2h),2.91(ddd,j=17.2,13.5,5.3hz,1h),2.66
–
2.55(m,1h),2.36(dt,j=17.1,7.2hz,4h),2.07
–
1.98(m,0h),1.66(td,j=7.4,2.9hz,4h),1.39(h,j=7.6,6.5hz,2h).
13
c nmr(151mhz,dmso-d6)δ172.27,170.76,170.66,170.49,167.25,163.14,161.88,143.63,143.15,140.46,139.35,133.22,133.06,132.07,128.63,128.01,124.62,119.61,118.35,112.20,111.36,107.47,104.89,54.31,50.93,45.89,35.71,35.08,30.59,27.71,24.32(d,j=5.4hz),22.03.hrms(esi,m/z)calcd for c
32h31
n7o5s2[m+na]
+
,680.1828;found,680.1736
[0069][0070]
化合物p11、p12、p13、p14、p15的合成方法与p1的合成相同,仅将靶蛋白配体l1变为l3,与相应的1b-f反应得到。
[0071][0072]
p11:1h nmr(600mhz,dmso-d6)δ11.02(s,1h),10.62(s,1h),9.99(s,1h),9.76(s,1h),8.00(s,1h),7.95(d,j=8.5hz,2h),7.81(dd,j=7.4,1.6hz,1h),7.74
–
7.65(m,4h),7.60(s,1h),7.51
–
7.46(m,2h),7.42(d,j=8.8hz,2h),5.15(dd,j=13.3,5.2hz,1h),4.36(q,j=17.4hz,2h),3.00
–
2.85(m,1h),2.61(dt,j=17.1,3.5hz,1h),2.33(dt,j=18.1,7.3hz,5h),2.10
–
1.93(m,2h),1.60(dt,j=8.2,3.8hz,4h),1.23(s,1h).
13
c nmr(151mhz,dmso-d6)δ173.36,171.87,171.57,168.32,163.75,162.52,155.59,144.58,140.20,139.81,134.29,134.17,133.14,129.41,129.31,129.10,127.00,125.71,125.52,119.54,119.45,118.97,113.72,106.75,51.99,46.93,36.92,36.28,31.67,30.01
–
28.47(m),25.56,23.12.hrms(esi,m/z)calcd for c
38h34
cln7o5s2[m+h]
+
,768.1751;found,768.1816.
[0073][0074]
化合物3a的合成
[0075]
将庚二酸(175mg,1.09mmol)和socl2(80μl,1.09mmol)置于50ml反应瓶中,加入thf 10ml,30min后,减压浓缩除去thf和多余的socl2,加入泊马度胺(化合物2,300mg,1.09mmol),diea,thf 15ml,70℃下搅拌3h,tlc显示反应完全。加水30ml,ea萃取3次(20ml*3),合并有机层,柱色谱分离,得到淡黄色油状物3a 210mg,收率:46.4%。
[0076]
化合物3b合成方法与3a相同。
[0077]
化合物p16的合成
[0078][0079]
按照p1的合成方法,将3a和l3按照1:1的比例合成,得到化合物p16 40mg,收率:43.8%。1h nmr(600mhz,dmso-d6)δ11.08(s,4h),7.90(s,3h),7.62(t,j=7.7hz,4h),7.38(d,j=7.1hz,3h),7.26(d,j=8.2hz,3h),4.22(t,j=6.6hz,1h),2.90(d,j=5.3hz,1h),2.88(d,j=2.3hz,1h),2.85(d,j=5.3hz,1h),2.60(s,1h),2.58
–
2.57(m,1h),2.53(d,j=5.1hz,1h),2.01(d,j=2.5hz,1h),2.01
–
1.98(m,2h),1.64(p,j=6.8hz,1h),1.37(q,j=7.5hz,1h),1.23(s,2h),0.91(t,j=7.4hz,1h).
13
c nmr(151mhz,dmso-d6)δ173.20,170.40,167.49,156.79,152.23,135.27,133.29,130.20,118.99,116.37,48.98,34.32,31.33,22.47.
[0080]
化合物p17的合成
[0081][0082]
按照p1的合成方法,将3b和l3按照1:1的比例合成,得到化合物p17 52mg,收率:44.2%。
[0083]
化合物4的合成
[0084]
将化合物2(300mg,1.09mmol),氯乙酰氯(104μl,1.31mmol),thf 15ml,加入到
50ml反应瓶内,60℃反应2h,待反应液冷却,抽滤,得到白色固体268mg,收率:70.6%。
[0085]
化合物5的合成
[0086]
将化合物4(200mg,0.57mmol),n-boc哌嗪(106mg,0.57mmol),diea(153μl,0.86mmol),碘化钾10mg加入25ml反应瓶中,加入8ml dmf,80℃反应过夜,tlc显示反应完全,将反应液倒入30ml水中,ea萃取3次(20ml*3),合并有机层,无水硫酸钠干燥。加入2m hcl/ea 5ml,搅拌2h,直接抽滤得到淡黄色盐酸盐形态产品200mg,收率:80.3%。
[0087]
化合物6a的合成
[0088]
将l3(150mg,0.39mmol),6-溴己酸(98mg,0.47mmol),hatu(178mg,0.47mmol),diea(84μl,0.47mmol),加入25ml反应瓶中,加入8ml dmf,室温搅拌2h,tlc监测反应完全,反应液倒入30ml水中,ea萃取3次(20ml*3),合并有机层,将反应液减压浓缩得到黄色油状物中间体化合物6a,收率:96.1%。
[0089]
化合物6b、6c、6d、6e、6f、6g按相同方法合成。
[0090]
化合物p18合成
[0091][0092]
将化合物6a(200mg,0.36mmol),化合物5(144mg,0.36mmol),diea(84μl,0.47mmol),碘化钾10mg加入25ml反应瓶中,加入8ml dmf,80℃反应过夜,tlc显示反应完全,将反应液倒入30ml水中,ea萃取3次(20ml*3),合并有机层,无水硫酸钠干燥。柱层析得到目标化合物220mg,收率:69.4%。1h nmr(600mhz,dmso-d6)δ11.15(s,1h),10.96(s,1h),10.64(s,1h),10.01(s,1h),8.78(d,j=8.5hz,1h),8.00(s,1h),7.95(d,j=8.3hz,2h),7.84(t,j=7.9hz,1h),7.71(dd,j=15.1,8.5hz,4h),7.60(s,1h),7.59(d,j=7.3hz,1h),7.41(d,j=8.5hz,2h),5.16(dd,j=12.8,5.5hz,1h),3.27
–
3.09(m,2h),2.99
–
2.86(m,1h),2.65
–
2.51(m,8h),2.34(t,j=7.4hz,4h),2.10(ddd,j=10.7,6.5,3.8hz,1h),1.62(t,j=7.5hz,2h),1.55
–
1.43(m,2h),1.33(t,j=7.5hz,2h),1.22(s,2h).
13
c nmr(151mhz,dmso-d6)δ173.28,171.83,170.60,170.37,168.42,167.30,163.74,162.52,155.59,144.57,140.20,139.81,136.83,131.84,129.39,127.00,125.51,124.69,119.54,118.97,118.40,116.22,113.72,106.75,61.87,58.06,53.44,52.98,49.38,36.90,33.98,32.23,31.38,29.64
–
28.90(m),27.04,26.61,25.53,24.23,22.41.hrms(esi,m/z)calcd for c
43h42
cln9o6s2[m+h]
+
,880.2388;found,880.2505
[0093]
化合物p19、p20、p21、p22、p23、p24按相同方法合成。
[0094][0095]
化合物8的合成
[0096]
将化合物7(200mg,0.72mmol),叔丁基4-(2-氨基乙基)哌嗪-1-羧酸酯(135mg,0.72mmol),diea(194μl,1.08mmol),加入25ml反应瓶中,加入10ml dmf,80℃反应过夜,tlc显示反应完全,将反应液倒入30ml水中,ea萃取3次(30ml*3),合并有机层,无水硫酸钠干燥。加入2m hcl/ea5ml,搅拌2h,直接抽滤得到淡黄色盐酸盐形态产品218mg,收率:77.9%。
[0097]
化合物p25的合成
[0098][0099]
将化合物6a(200mg,0.36mmol),化合物8(138mg,0.36mmol),diea(84μl,0.47mmol),碘化钾10mg加入25ml反应瓶中,加入8ml dmf,80℃反应过夜,tlc显示反应完全,将反应液倒入30ml水中,ea萃取3次(20ml*3),合并有机层,无水硫酸钠干燥。柱层析得到目标化合物207mg,收率:66.7%。1h nmr(600mhz,dmso-d6)δ11.09(s,1h),10.63(s,1h),10.00(s,1h),8.00(s,1h),7.95(d,j=8.5hz,2h),7.71(dd,j=16.5,8.7hz,4h),7.60(s,1h),7.57(dd,j=8.5,7.1hz,1h),7.41(d,j=8.9hz,2h),7.07(d,j=8.6hz,1h),7.02(d,j=7.0hz,1h),6.73(t,j=5.2hz,1h),5.06(dd,j=12.9,5.4hz,1h),2.88(ddd,j=17.0,13.8,5.5hz,1h),2.60(q,j=2.9hz,1h),2.55(t,j=6.3hz,2h),2.46
–
2.36(m,4h),2.33(t,j=7.4hz,3h),2.26(t,j=7.2hz,3h),2.06
–
1.98(m,1h),1.65
–
1.58(m,2h),1.45(p,j=7.4hz,2h),1.36
–
1.28(m,3h),1.23(d,j=5.2hz,3h),0.85(t,j=6.9hz,1h).
13
c nmr(151mhz,dmso-d6)δ173.30,171.83,170.60,169.33,167.84,163.76,162.52,155.60,146.73,144.58,140.21,139.82,136.73,132.60,130.13,129.40,129.32,127.00,125.52,119.55,118.98,117.93,113.71,110.92,109.64,106.75,58.22,56.38(d,j=20.5hz),53.35(d,j=5.2hz),53.04(d,j=8.9hz),49.00,39.19(d,j=13.0hz),36.91,31.46,29.50(t,j=7.7hz),29.32(d,j=5.6hz),29.19(d,j=6.8hz),29.05,27.04(d,j=
5.7hz),26.62,25.56(d,j=6.3hz),22.58(d,j=6.5hz).hrms(esi,m/z)calcd for c
43h44
cln9o5s2[m+h]
+
,866.2595;found,866.2649
[0100]
化合物p26、p27、p28、p29按相同方法合成。
[0101][0102][0103]
化合物10a、10b、10c、10d、10e、10f、10g的合成与化合物6a合成方法相同,以化合物9和不同长度的溴代脂肪酸按1:1条件反应,得到相应目标化合物。
[0104]
化合物p30的合成
[0105][0106]
在50ml反应瓶中,加入化合物10a(320mg,0.52mmol),化合物l4(200mg,0.52mmol),cs2co3(400mg,1.02mmol),加入乙腈15ml,80℃反应6h,tlc显示反应完全,将反应液倒入40ml水中,ea萃取3次(30ml*3),合并有机层,无水硫酸钠干燥。柱色谱分离,得到目标化合物淡黄色固体167mg,收率:34.6%。1h nmr(400mhz,chloroform-d)δ11.70(s,1h),8.59(s,1h),7.56(dd,j=7.9,1.6hz,1h),7.45(s,1h),7.38
–
7.33(m,2h),7.27(d,j=3.6hz,3h),7.24(d,j=8.6hz,2h),7.19(s,1h),7.18
–
7.13(m,1h),7.08(s,1h),6.95(dd,j=8.3,1.2hz,1h),6.82(td,j=7.5,1.3hz,1h),6.01(d,j=8.6hz,1h),4.99(p,j=7.0hz,1h),4.63(t,j=7.8hz,1h),4.49
–
4.41(m,1h),4.37(d,j=8.6hz,1h),4.05
–
3.95(m,1h),3.90(t,j=7.3hz,2h),3.44(dd,j=11.4,3.6hz,1h),2.51(ddd,j=13.4,7.5,4.7hz,1h),2.42(s,3h),2.15(t,j=7.5hz,2h),2.08
–
1.92(m,1h),1.62(dp,j=15.2,7.5hz,5h),1.43(d,j=6.9hz,3h),1.38
–
1.25(m,2h),1.18(s,1h),0.74(s,9h).
13
c nmr(101mhz,chloroform-d)δ173.36,171.85,169.65,169.20,162.43,155.89,154.19,150.07,148.17,144.07,142.75,132.96,131.43,130.58,130.18,129.91,129.29,128.03,126.46,125.93,119.20,117.62,117.23,111.79,105.52,69.78,58.13,57.27,56.35,52.58,48.65,35.97,35.15,34.33,29.46,27.03,26.08,25.97,24.82,21.67,15.80.hrms(esi,m/z)calcd for c
47h52
cln7o5s2[m+h]
+
,926.2881;found,926.2953
[0107]
化合物p31-p36合成与p30相同。
[0108][0109]
化合物p37-p43的合成与p30相同,仅将靶蛋白配体由l4变为l5即可。
[0110]
[0111][0112]
化合物11的合成
[0113]
将化合物9(300mg,0.67mmol),氯乙酰氯(64μl,0.81mmol),diea(180μl,1.00mmol),thf 15ml,加入到50ml反应瓶内,60℃反应2h,待反应液冷却,将反应液倒入40ml水中,ea萃取3次(30ml*3),合并有机层,无水硫酸钠干燥。减压浓缩除去ea,得到黄色油状物,未经进一步处理直接进行后续反应。
[0114]
化合物12的合成
[0115]
将化合物11(598mg,1.15mmol),nan3(0.21g,3.30mmol),dmf 8ml加入到反应瓶内,6h后,tlc显示反应完全,将反应液倒入40ml水中,ea萃取3次(30ml*3),合并有机层,柱色谱分离,得到目标化合物515mg,收率:85%。
[0116]
化合物l5的合成
[0117]
将l4(300mg,0.78mmol),7-溴-1-庚炔(101mg,0.78mmol),cs2co3(600mg,1.55mmol),加入到50ml反应瓶,加入乙腈15ml,80℃反应6h,tlc显示反应完全,将反应液倒入40ml水中,ea萃取3次(30ml*3),合并有机层,无水硫酸钠干燥。柱色谱分离,得到目标化合物淡黄色固体186mg,收率:49.8%。
[0118]
化合物l6的合成方法与l5相同。
[0119]
化合物p44的合成
[0120][0121]
将化合物l5(170mg,0.32mmol),化合物12(153mg,0.32mmol),cuso4(32mg,0.12mmol),异抗坏血酸钠(51mg,0.25mmol),t-buoh/h2o(10ml,1:1)加入50ml反应瓶进行click反应,柱色谱分离,得到目标化合物169mg,收率:52.5%。1h nmr(600mhz,dmso-d6)δ10.84(s,1h),8.99(s,1h),8.56(d,j=7.4hz,1h),8.51(d,j=9.2hz,1h),8.16(s,1h),8.07(d,j=7.7hz,1h),7.79(s,1h),7.55(d,j=19.0hz,6h),7.47(s,1h),7.37(d,j=7.8hz,2h),7.19(d,j=7.9hz,1h),7.04(d,j=8.1hz,1h),6.90(t,j=7.7hz,1h),5.28
–
5.11(m,3h),4.91
–
4.80(m,1h),4.51(d,j=8.9hz,2h),4.35(s,1h),3.98(t,j=7.3hz,2h),3.71
–
3.51(m,2h),2.48(d,j=33.3hz,5h),2.04(t,j=10.3hz,1h),1.92(d,j=10.4hz,1h),1.66(dt,j=16.3,7.9hz,4h),1.39(d,j=7.2hz,5h),0.90(s,9h).
13
c nmr(151mhz,dmso-d6)δ171.68,169.75,169.44,165.95,161.54,155.65,152.77,151.94,148.08,146.86,144.76,144.65,143.38,131.95,131.75,130.58,129.82,129.64,128.96(d,j=4.8hz),127.10,123.90,119.88,119.63,117.13,116.72,106.26,69.34,59.20,57.30,56.94,52.72,51.71,48.09,38.32,36.04,29.01,27.18,26.72,26.10,25.31,22.55,16.41.hrms(esi,m/z)calcd for c
50h55
cln
10
o5s3[m+na]
+
,1029.3208;found,1029.3195.
[0122]
化合物p45的合成与p44方法相同。
[0123][0124]
p45:1h nmr(600mhz,dmso-d6)δ10.83(s,1h),8.98(s,1h),8.56(dd,j=11.4,8.4hz,
[0125]
2h),8.17(s,1h),8.07(dd,j=7.8,1.8hz,1h),8.02(s,1h),7.58(d,j=8.7hz,2h),7.56
–
7.52(m,4h),7.47(s,1h),7.36(d,j=8.1hz,2h),7.18(td,j=7.6,1.8hz,1h),7.03(d,j=8.0hz,1h),6.90(t,j=7.3hz,1h),5.24(d,j=4.8hz,2h),5.20(d,j=3.4hz,1h),4.86(t,j=7.2hz,1h),4.51(d,j=5.1hz,2h),4.50(s,2h),4.34(dp,j=6.6,3.6hz,1h),4.11(t,j=5.6hz,2h),3.70(t,j=5.6hz,2h),3.64(dd,j=10.5,4.1hz,1h),3.55(d,j=10.0hz,1h),3.54
–
3.49(m,4h),2.45(s,3h),2.03(dd,j=12.5,7.5hz,1h),1.90(ddd,j=13.0,8.4,4.6hz,1h),1.39(d,j=7.0hz,3h),0.90(s,9h).
13
c nmr(151mhz,dmso-d6)δ171.69,169.72,169.44,165.80,161.47,155.65,152.77,151.95,148.09,144.76,144.65,
144.00,143.69,132.21,131.75,130.50,129.82,129.65,129.46,128.94,128.82,127.10,126.06,119.89,119.63,117.11,116.71,106.50,70.01,69.34,67.49,63.91,59.20,57.35,56.97,52.55,51.74,48.09,38.30,36.03,26.72,22.56,16.41.hrms(esi,m/z)calcd for c
50h55
cln
10
o7s3[m+na]
+
,1061.3106;found,1061.3014.
[0126]
其中化合物p31的1h nmr光谱和
13
c nmr光谱分别如图1、图2所示。
[0127]
实施例2
[0128]
上述实施例1中制得的protac化合物在蛋白免疫印迹实验水平的活性筛选
[0129]
实验方法:
[0130]
细胞处理:
[0131]
(1)取对数生长期a549/taxol细胞按3
×
105密度接种,待细胞贴壁后使用实施例1中制得的protac化合物处理,孵育相应时间后收细胞。
[0132]
(2)吸出6孔板中的培养液,残存的培养液用4℃预冷的磷酸盐缓冲液(pbs)清洗,并将残留的pbs吸净。在冰上向每个孔中加入60μl已加入pmsf的ripa裂解液后,用细胞刮刀刮取蛋白并收集至2ml ep管中。待所有样品刮取完成后,转移至低温高速离心机中4℃,15000rpm离心20min,结束后吸取蛋白上清转移至1.5ml离心管中,随后进行蛋白定量或-80℃冻存备用。蛋白定量后,按蛋白上样量40μg计算上样体积。
[0133]
western blot检测的具体步骤如下:
[0134]
(1)配制合适浓度的sds-page胶。制备浓度为10%的分离胶。
[0135]
(2)制备样品。蛋白样品中加入对应体积的5
×
溴酚蓝上样缓冲液(使其终浓度为1
×
)及5%β-巯基乙醇并混匀,100℃水浴5min后,-80℃保存备用。
[0136]
(3)电泳。接通电源,蛋白样品在浓缩胶中电压为70伏特,待蛋白样品进入分离胶时,我们把电压调整为110伏特继续电泳。待溴酚蓝几乎完全跑出分离胶时终止电泳。
[0137]
(4)转膜。电泳结束后取下凝胶,按下列顺序安装转膜装置:(负极)、转膜海绵、滤纸、凝胶、活化的pvdf膜、滤纸、转膜海绵、(正极)。切记凝胶和pvdf膜之间绝对不能气泡。然后夹紧转移装置置于转膜缓冲液中,最后放入冰盒,以25v电转0.5h后54v电转1.5h。
[0138]
(5)封闭。转膜结束后,取出pvdf膜,将膜浸没在含5%的脱脂奶粉的tbst缓冲液里,室温下摇床缓慢振荡1h。
[0139]
(6)一抗孵育。封闭结束后,加入适度稀释比例的一抗,4℃过夜。回收一抗,将pvdf膜用tbst缓冲液荡洗3次,每次振荡10min。
[0140]
(7)二抗孵育。弃去tbst缓冲液,加入一定稀释比(通常是1:7500)的二抗(鼠抗或者兔抗,由一抗决定),室温下摇床缓慢振荡1h。弃去二抗,将pvdf膜用tbst缓冲液荡洗3次,每次振荡10min。最后用tbst缓冲液荡洗10min。
[0141]
(8)显色并压片。将ecl显色底物均匀覆盖在pvdf膜上,室温显色。
[0142]
实验结果:
[0143]
发明人应用免疫印迹法对部分protac化合物分子降解cyp1b1的能力进行了评估,在紫杉醇耐药的非小细胞肺癌细胞系a549/taxol内进行免疫印迹分析,结果显示:cyp1b1的配体n-(4-氯苯基)-4-苯基-[2,4
′‑
联噻唑]-2
′‑
胺(pro-1)(cn114249702a n-芳基-[2,4
′‑
双噻唑]-2
′‑
胺类化合物及其制备与用途)不具有降解cyp1b1活性,而本发明提供的化合物p30、p31、p32、p39、p42、p43展现了降解活性。但是化合物p31在1nm时即可降解cyp1b1
达50%,1000nm时仍然具有较好的降解效果。其他降解剂在1nm时有一定降解效果,但是在1000nm因为hook效应,几乎不具有降解效果(图3),因此选择化合物p31进行浓度依赖性和时间依赖性考察。
[0144]
本发明检测了不同浓度化合物p31加入a549/taxol细胞内作用后化合物p31对cyp1b1蛋白的降解情况,如图4所示:在24h的孵育后,在a549/taxol细胞中,化合物p31在0.1nm展现出》50%的降解效果,dc
50
(half maximal degradation concentration)约为0.1nm,其中1nm和10nm降解效果最优,达到了80%以上。给药浓度至100nm时,由于hook效应存在,降解能力下降,至10μm时,则失去降解能力。选取化合物p31浓度为10nm,与a549/taxol细胞分别孵育1h,6h,12h,24h,36h,48h后,考察cyp1b1的降解效果,结果发现,在1h时开始降解cyp1b1蛋白,24h后,降解效果达到峰值,可降解cyp1b1蛋白80%以上。
[0145]
基于上述结果,发明人继续考察了化合物p31是否通过cyp1b1-protac-vhl三元复合物及泛素蛋白酶体依赖的降解方式,本发明使用cyp1b1抑制剂pro-1,vhle3连接酶的抑制剂vh032,蛋白酶体抑制剂mg132,以及不具备vhle3连接酶亲和力的阴性分析化合物p46,预处理细胞2小时,再加化合物p31处理后,最后通过wb检测cyp1b1蛋白条带的变化结果。如图5所示:上述处理均能逆转p31对cyp1b1的降解作用,且阴性配体p46也无降解效果,表明降解剂p31确实通过cyp1b1-protac-vhl三元复合物及泛素蛋白酶体依赖的降解方式降解cyp1b1。
[0146]
实施例3
[0147]
实施例化合物p31的逆转耐药性测试
[0148]
实验原理:mtt分析法是一种以活细胞代谢还原四甲基偶氮哇盐(3-(4,5-dimcthyl-2thiahiazoyl)-3,5-di-phenyl-tetrazolium bromide,mtt)为基础的检测细胞存活率的实验方法。由于外源性加入的mtt能被活细胞线粒体中的琥珀酸脱氢酶还原成不溶于水的蓝紫色结晶-甲瓒,并沉积在细胞中,然而在死细胞中并无此功能。甲瓒结晶能被dmso溶解,在酶标仪492nm处有最大吸收,通过检测吸光度值的高低,可以间接反映活细胞数目。
[0149]
实验步骤:取对数生长期的a549/taxol细胞以3.0
×
103细胞/孔接种于96孔细胞培养板,培养过夜。待细胞贴壁稳定后,次日吸出原培养液,分别加入100μl含不同浓度、不同组合的待测物溶液,每组设置至少3个复孔。置于37℃、5%co2细胞培养箱中培养72h后弃去上清液,每孔加入100μl pbs洗涤1次,然后每孔加入100μl 0.5mg/ml的mtt,培养3-5h后除去mtt,每孔加入100μl dmso,震荡3min使甲瓒结晶溶解,于酶标仪492nm处检测其吸光度,间接反映活细胞数量。记录数据。
[0150]
实验结果:
[0151]
发明人根据不同浓度的化合物p31与不同浓度的紫杉醇联用,考察对450泰素耐药细胞株a549/taxol的存活率影响。如图6所示:0.1nm,1nm,10nm,的化合物p31能与500nm,1000nm,5000nm的紫衫醇具有较强的联合作用,有效的杀伤肿瘤细胞的存活率,呈现浓度依赖性,表明实施例化合物p31降解cyp1b1后可以有效的逆转耐药细胞株对于紫杉醇的耐药作用。
[0152]
实施例4
[0153]
实施例化合物p31进行细胞迁移侵袭实验。
[0154]
实验方法:
[0155]
划痕实验:
[0156]
测定原理:细胞划痕法是一种简捷测定细胞迁移运动与修复能力的方法,类似于体外伤口愈合模型。在体外培养皿或平板中培养的单层贴壁细胞层,用微量枪头在细胞生长的中央区域划线,所划区域称为“划痕”。划痕边缘的细胞会逐渐进入中央划痕区,使“划痕”愈合。去除中央部位的细胞后继续培养细胞至实验预先设定的时间,通过观察细胞是否迁移至中央划痕区来判断细胞的迁移能力。细胞迁移能力越强,其中央划痕区域剩余面积越小。
[0157]
实验步骤:
[0158]
(1)取对数生长期的细胞,以5.0
×
105细胞/孔的密度接种于6孔板中,培养箱中培养24h;
[0159]
(2)待细胞贴壁后,用200μl的黄色枪头垂直于6孔板沿中间区域划痕;
[0160]
(3)用pbs清洗细胞3次,洗掉划下来的细胞,按照预先设计的给药方案给药,放入培养箱中培养,然后分别在0h、24h时使用倒置显微镜观察并拍照。用image j处理定量。
[0161]
transwell实验
[0162]
实验原理:transwell实验的主要材料是transwell小室,即可置于细胞培养板里的小杯子,杯子底部有一层具有一定孔径的通透性聚碳酸酯膜。将transwell(8μm小室)小室置于24孔板中,小室内称为上室,培养板内称为下室,上下层培养液以膜相隔,并在膜上室铺基质胶,用来模拟体内细胞外基质,下室铺明胶。实验将细胞接种于上室,由于聚碳酸酯膜有通透性,下层培养液中的成分可以影响到上室内的细胞,如果上室的细胞要转移到下室,还需要分泌基质金属蛋白酶将基质胶降解,才能通过聚碳酸酯膜,最后通过计数下室明胶上粘住的细胞数即可反映细胞的侵袭能力。
[0163]
实验步骤:
[0164]
(1)transwell小室的制备。提前将高浓度基质胶置于冰中放在4℃溶解;
[0165]
(2)将溶解的高浓度基质胶用预冷的pbs稀释30倍,然后加入40μl稀释好的基质胶到每个小室的上室中,摇晃均匀,待均匀铺展后吸出15μl,注意不能有气泡,不能接触到膜,放在生物安全柜中晾干,即上室包被完成。随后进行下室非细胞面包被,吸取20μl 0.2%明胶(使用前需要过滤除菌)涂抹均匀,用黄枪头倾斜将其涂匀,晾干,即下室包被完成;
[0166]
(3)取对数生长期细胞,按照3.5
×
105细胞/孔的密度接种于小室的上室中,其中上室为200μl含细胞的0.5%fbs培养液,下室为600μl含20%fbs的培养液。铺板时按照给药方案给药,放入37℃、5%co2培养箱中培养;
[0167]
(4)培养24小时后,取出小室,用pbs洗涤上室2次,然后用棉签轻柔擦掉上室的细胞,使用4%多聚甲醛固定20min;
[0168]
(5)固定完毕后使用1%结晶紫染液染色30min;
[0169]
(6)用镊子夹住小室于清水中反复冲洗,直至小室内无明显结晶紫残留;
[0170]
(7)自然晾干后,使用倒置显微镜对小室底部明胶上面粘附的染色细胞进行拍照记录。
[0171]
实验结果:
[0172]
cyp1b1蛋白已经被证实与肿瘤细胞株的迁移侵袭密切相关,发明人在化合物p31
可降解cyp1b1蛋白的基础上考察化合物p31对a549/taxol细胞株的迁移侵袭作用。如图7所示:在可降解cyp1b1的浓度下(0.1nm,1nm,10nm),化合物p31能显著抑制a549/taxol细胞株的迁移作用,同样的,在上述浓度下,化合物p31也能抑制a549/taxol细胞株的侵袭作用,以上结果说明,该降解剂可以因降解cyp1b1后抑制肿瘤细胞株a549/taxol的迁移侵袭作用。
[0173]
因此,总结起来,发明人通过western blot实验证实这一类降解剂对cyp1b1的降解效果。进一步通过与泰素细联合给药的细胞增殖实验、划痕实验以及transwell实验证实证明这一类cyp1b1降解剂可以有效抑制细胞对紫杉醇的耐药作用,以及迁移侵袭作用。
[0174]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1