一种2-乙酰基噻唑的制备方法与流程
1.本发明属于食品香料合成领域,特别涉及一种2-乙酰基噻唑的制备方法。
背景技术:
2.公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
3.2-乙酰基噻唑主要应用在食品香料行业,是一种常见的食用香料,其天然存在于熟牛肉、熟猪肝、土豆、米饭中,具有类似坚果、稻谷、爆米花的香味,广泛应用于食品添加剂中。
4.该化合物常见的合成方法主要有两种。其中最常用,也是工业生产中使用的方法主要是利用2-溴噻唑在丁基锂在-78℃左右发生锂溴交换,后与乙酸乙酯或者乙酰胺反应得到2-乙酰基噻唑。另一种办法是将2-溴噻唑与三甲基氯硅烷先反应,制得2-三甲基硅基噻唑,再与乙醛作用得到2-乙酰基噻唑。还有文献提到过利用正噻唑与乙酰氯在三乙胺存在下直接酰化得到2-乙酰基噻唑,但该反应收率仅为3%。
5.目前已知的主要合成方法都存在一定的问题,丁基锂的工艺需要特殊的反应温度,不利于生产放大;三甲基氯硅烷的工艺中原料成本较高,操作复杂,导致工艺成本太高,不具备应用前景。
技术实现要素:
6.为了解决上述问题,本发明提供一种全新的合成路线,更加适宜进行工业化生产的制备方法。本路线避免使用安全性差的丁基锂试剂,同时无需-78℃的超低温,以便宜易得的2-甲磺酰基噻唑为起始原料,在碱的作用下与2-甲基-1,3-二噻烷反应,最后水解、中和后处理可得到产品2-乙酰基噻唑。
7.为了实现上述目的,本发明采用如下技术方案:
8.本发明的第一个方面,提供了一种2-乙酰基噻唑的制备方法,包括:
9.将2-甲磺酰基噻唑、2-甲基-1,3-二噻烷分散于溶剂中,在碱存在的条件下,进行反应,反应完毕后,降温、加酸进行反应,反应完成后,调节体系ph值至7~7.5之间,分出有机相,浓缩,即得。
10.发明人在噻唑类化合物的合成研究中,发现了一新的合成方法,技术路线如下:
[0011][0012]
并利用该方法成功的制备了2-乙酰基噻唑,该工艺条件温和,原料便宜易得,具有工业化应用的前景。
[0013]
本发明的第二个方面,提供了上述的方法制备的2-乙酰基噻唑。
[0014]
本发明的有益效果
[0015]
(1)相比较于目前已知的2-乙酰基噻唑合成方法,本发明所使用的方法反应条件温和,收率可观,避免了使用价格较高的丁基锂溶液,也避免了苛刻的低温反应条件,具备进行工业化放大生产的前景。
[0016]
(2)本发明制备方法简单、实用性强,易于推广。
附图说明
[0017]
构成本技术的一部分的说明书附图用来提供对本技术的进一步理解,本技术的示意性实施例及其说明用于解释本技术,并不构成对本技术的不当限定。
[0018]
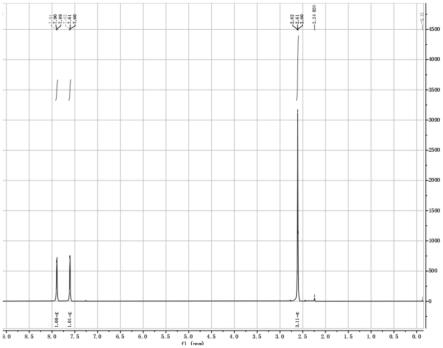
图1为本发明实施例1制备的2-乙酰基噻唑核磁氢谱。
具体实施方式
[0019]
应该指出,以下详细说明都是例示性的,旨在对本技术提供进一步的说明。除非另有指明,本发明使用的所有技术和科学术语具有与本技术所属技术领域的普通技术人员通常理解的相同含义。
[0020]
一条新的2-乙酰基噻唑的合成方法,包括:
[0021]
室温下,将2-甲磺酰基噻唑、2-甲基-1,3-二噻烷分散于thf中,搅拌溶解,加入碱,加毕,升温,体系内温在55~60℃反应6小时;转移至冰水浴中,向体系中滴加20%的稀硫酸溶液,滴毕,于室温搅拌反应1h;加入5%的碳酸氢钠溶液,调节体系ph值至7~7.5之间,分出有机相,浓缩得烷基噻唑粗品,精馏得纯品。
[0022]
在一些实施例中,所述2-甲磺酰基噻唑、2-甲基-1,3-二噻烷的质量比为16.3~18:14~16。
[0023]
在一些实施例中,所述碱为氢氧化钾、氢氧化钠、氢化钠、叔丁醇钾中至少一种。
[0024]
在一些实施例中,所述碱与2-甲基-1,3-二噻烷的质量比为5~15:14。
[0025]
在一些实施例中,所述2-甲磺酰基噻唑、2-甲基-1,3-二噻烷的反应温度为55~60℃,反应时间为6~8h。
[0026]
在一些实施例中,降温至12~16℃。
[0027]
在一些实施例中,酸的浓度为20~30%,体积为溶剂体积的1/4~1/3。
[0028]
在一些实施例中,加酸进行反应的时间为1~1.5h。
[0029]
下面结合具体的实施例,对本发明做进一步的详细说明,应该指出,所述具体实施例是对本发明的解释而不是限定。
[0030]
实施例1
[0031]
2-乙酰基噻唑的制备
[0032]
将16.3g 2-甲磺酰基噻唑、14g 2-甲基-1,3-二噻烷依次加入到200ml thf中,搅拌溶解。于室温下,加入8g氢氧化钾,开启加热,保持内温为55~60℃之间,保温反应6h;
[0033]
将上述体系降温至15℃左右,滴加50ml 20%的硫酸溶液,控制滴加速度,保持内温25℃以内,加毕,升至室温搅拌1h;滴加5%的碳酸氢钠溶液,调节ph至7~7.5之间,分出有机相,水相再用200ml thf萃取一次,合成有机相,浓缩得2-乙酰基噻唑粗品,粗品精馏纯
化得2-乙酰基噻唑纯品9.2g,收率72%。1h-nmr(dmso-d6):δ=2.61(s,3h),7.60~7.62(d,1h),7.89~7.91(d,1h).
[0034]
实施例2
[0035]
2-乙酰基噻唑的制备
[0036]
将16.3g 2-甲磺酰基噻唑、14g 2-甲基-1,3-二噻烷依次加入到200ml thf中,搅拌溶解。于室温下,加入6g氢氧化钠,开启加热,保持内温为55~60℃之间,保温反应6h;
[0037]
将上述体系降温至15℃左右,滴加50ml 20%的硫酸溶液,控制滴加速度,保持内温25℃以内,加毕,升至室温搅拌1h;滴加5%的碳酸氢钠溶液,调节ph至7~7.5之间,分出有机相,水相再用200ml thf萃取一次,合成有机相,浓缩得2-乙酰基噻唑粗品,粗品精馏纯化得2-乙酰基噻唑纯品7.4g,收率58%。
[0038]
实施例3
[0039]
2-乙酰基噻唑的制备
[0040]
将16.3g 2-甲磺酰基噻唑、14g 2-甲基-1,3-二噻烷依次加入到200ml thf中,搅拌溶解。于室温下,加入5g氢化钠(60%),开启加热,保持内温为55~60℃之间,保温反应6h;
[0041]
将上述体系降温至15℃左右,滴加50ml 20%的硫酸溶液,控制滴加速度,保持内温25℃以内,加毕,升至室温搅拌1h;滴加5%的碳酸氢钠溶液,调节ph至7~7.5之间,分出有机相,水相再用200ml thf萃取一次,合成有机相,浓缩得2-乙酰基噻唑粗品,粗品精馏纯化得2-乙酰基噻唑纯品10.5g,收率82.6%。
[0042]
实施例4
[0043]
2-乙酰基噻唑的制备
[0044]
将16.3g 2-甲磺酰基噻唑、14g 2-甲基-1,3-二噻烷依次加入到200ml thf中,搅拌溶解。于室温下,加入15g叔丁醇钾,开启加热,保持内温为55~60℃之间,保温反应6h;
[0045]
将上述体系降温至15℃左右,滴加50ml 20%的硫酸溶液,控制滴加速度,保持内温25℃以内,加毕,升至室温搅拌1h;滴加5%的碳酸氢钠溶液,调节ph至7~7.5之间,分出有机相,水相再用200ml thf萃取一次,合成有机相,浓缩得2-乙酰基噻唑粗品,粗品精馏纯化得2-乙酰基噻唑纯品11.5g,收率90.5%。
[0046]
以上所述仅为本技术的优选实施例而已,并不用于限制本技术,对于本领域的技术人员来说,本技术可以有各种更改和变化。凡在本技术的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本技术的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1