一种促进骨再生或骨形成的多肽的制备及用途
1.本发明涉及生物医药技术领域,特别是涉及一种促进骨再生或骨形成的多肽的制备及用途。
背景技术:
2.先天疾病、癌症、创伤、感染等原因引起的口腔颌面部骨缺损严重影响患者的生活质量。组织工程即干细胞、支架和生长因子的结合,为功能性骨再生带来了新的希望。多种生长因子参与调控骨再生过程的不同阶段;其中一些生长因子已被广泛应用于临床前修复骨缺损模型。主要包括:骨形态发生蛋白(bone morphogenetic proteins,bmps),血管内皮生长因子-a,血小板衍生生长因子-bb,成纤维细胞生长因子-2等。其中,骨形态发生蛋白(bone morphogenetic protein 2,bmp2),是应用最广泛的骨移植辅助治疗的生长因子。
3.bmps参与骨再生过程的所有阶段,包括软骨生成、血管生成和成骨。小鼠模型已经确定bmp2是启动骨修复反应所需的刺激,bmp2缺失会导致骨痂形成困难。美国食品和药物管理局批准bmp2用于腰椎融合、胫骨开放性骨折等,bmp7用于骨折修复、脊柱融合、以及用于加速愈合和降低感染率等。然而,bmps的释放动力学快、蛋白质稳定性差,需使用超生理剂量以维持足够高的局部浓度方可发挥作用,带来的潜在不良影响阻碍了其临床应用,如术后水肿、异位骨形成、骨移植吸收、骨溶解和增加癌症发展风险等。
技术实现要素:
4.针对现有技术的不足,本发明的目的是提供一种促进骨再生或骨形成的多肽的制备及用途。
5.本发明的目的是通过以下技术方案实现的:
6.第一方面,本发明提供了一种多肽在制备促进再生或骨形成的药物中的用途,所述多肽的氨基酸序列如seq id no.1所示。
7.本发明所述的多肽为具有促进bmp2诱导成骨功能的多肽,其来源于一种属于小富亮氨酸蛋白聚糖家族(small leucine-rich proteoglycans,slrps)的细胞外基质蛋白——骨调蛋白(osteomodulin,omd)。该家族特征是具有富亮氨酸重复序列(leucine-richrepeats,lrrs)的蛋白核心以及至少一个糖胺聚糖侧链。在功能上,slrps一方面可与生长因子结合形成特定浓度梯度来调节其生物利用度,另一方面介导细胞表面受体和生长因子结合,调节细胞与基质的相互作用,影响细胞功能。slrps在骨形成的所有阶段都扮演特定的角色,包括细胞生长、有机基质组装、矿物质沉积和改建。
8.本发明在前期的研究中发现,omd在体外可增强bmp2诱导的成骨相关基因的表达,且呈浓度依赖性;与单独加入omd或低浓度bmp2相比,同时使用omd和低浓度bmp2组在大鼠下颌骨缺损模型中可形成更成熟和丰富的矿化骨;omd可以通过其末端lrrs与bmp2结合,与bmp2及其膜受体bmpr形成复合物,从而促进bmp/smad 信号转导。omd是骨形成的正向调节因子,是bmp2信号的协调者,bmp2和omd 的结合显示出增强的生物活性。
9.omd共有421个氨基酸,在本发明中,我们定位了omd与bmp2相互作用关键结构域,选择性的缩短了该促进bmp2诱导成骨功能的蛋白的肽段长度,最终得到了本发明所述的一种促进bmp2诱导成骨功能的多肽。所述多肽为omd核心蛋白中11段 lrrs基序的第10-11段,omd通过第10-11个lrrs与bmp2结合,并与bmp受体形成复合物,从而激活下游smads调节转录反应,减少bmp2用量,优化bmp2成骨。本发明的多肽分子量小,结构简单且稳定,而且活性更高。
10.优选地,所述多肽的制备方法包括以下步骤:
11.a、构建过表达质粒:设计多肽的引物序列,进行pcr扩增获得目的基因,将含目的基因的pcr产物进行凝胶电泳,回收得到纯净的目的基因片段,将目的基因片段插入到酶切后的载体中得到重组质粒,再将重组质粒转化大肠杆菌,挑选单克隆细菌进行质粒抽提纯化,得过表达质粒;
12.b、获得多肽:将过表达质粒转化感受态大肠杆菌,挑选单克隆细菌接种lb培养基进行摇床培养,所得培养液中加入蛋白诱导剂进行处理,处理后的菌液经第一次离心、裂解后,再第二次离心、过滤,然后提取及去除内毒素后得到多肽。
13.优选地,步骤a中,所述多肽的引物序列如seq id no:14和seq id no:15所示;
14.所述载体为pgex-4t-1,酶切的位点为bamh i和not i;
15.所述大肠杆菌为感受态大肠杆菌,例如可以是top10,dh5α等大肠杆菌,但不限于此。
16.优选地,步骤b中,所述感受态大肠杆菌为感受态bl21;
17.所述lb培养基中含有氨苄青霉素;
18.所述摇床培养具体为:先37℃摇床培养6-7h,所得菌液再以1:20-200的体积比转接入新的lb培养基中,再37℃摇床培养至cd600值达到0.6。
19.优选地,步骤b中,所述蛋白诱导剂为浓度100μm-2mm的iptg;
20.所述处理的条件为:在20-30℃下处理12-14小时;
21.所述第一次离心的条件为4500-5500g、4-6℃、离心5-10min;
22.所述裂解的的条件为4-6℃裂解30-60min;
23.所述第二次离心的条件为11000-13000g、4-6℃、离心15-25min;
24.所述过滤采用0.22μm或0.45μm的滤膜。
25.优选地,所述方法还包括对步骤b获得的多肽进行去除内毒素的步骤。
26.优选地,所述去除内毒素采用的是高容量内毒素去除树脂。
27.优选地,所述促进骨损伤再生药物包括多肽和bmp2。
28.第二方面,本发明提供了一种促进骨再生或骨形成的多肽的制备方法,所述多肽的氨基酸序列如seq id no.1所示;
29.所述多肽的制备方法包括以下步骤:
30.a、构建过表达质粒:设计多肽的引物序列,进行pcr扩增获得目的基因,将含目的基因的pcr产物进行凝胶电泳,回收得到纯净的目的基因片段,将目的基因片段插入到酶切后的载体中得到重组质粒,再将重组质粒转化大肠杆菌,挑选单克隆细菌进行质粒抽提纯化,得过表达质粒;
31.b、获得多肽:将过表达质粒转化感受态大肠杆菌,挑选单克隆细菌接种lb培养基
进行摇床培养,所得培养液中加入蛋白诱导剂进行处理,处理后的菌液经第一次离心、裂解后,再第二次离心、过滤,然后提取及去除内毒素后得到多肽。
32.优选地,步骤a中,所述多肽的引物序列如seq id no:14和seq id no:15所示;
33.所述载体为pgex-4t-1,酶切的位点为bamh i和not i;
34.所述大肠杆菌为感受态大肠杆菌,例如可以是top10,dh5α等大肠杆菌,但不限于此。
35.优选地,步骤b中,所述感受态大肠杆菌为感受态bl21;
36.所述lb培养基中含有氨苄青霉素;
37.所述摇床培养具体为:先37℃摇床培养6-7h,所得菌液再以1:20-200的体积比转接入新的lb培养基中,再37℃摇床培养至cd600值达到0.6;
38.步骤b中,所述蛋白诱导剂为浓度100μm-2mm的iptg;
39.所述处理的条件为:在20-30℃下处理12-14小时;
40.所述第一次离心的条件为4500-5500g、4-6℃、离心5-10min;
41.所述裂解的的条件为6℃裂解30-60min;
42.所述第二次离心的条件为11000-13000g、4-6℃、离心15-25min;
43.所述过滤采用0.22μm或0.45μm的滤膜。
44.所述去除内毒素采用的是高容量内毒素去除树脂。
45.第三方面,本发明提供了一种促进骨损伤再生的药物,包括以下浓度的各组分: 0.1μg/ml bmp2、0.2-5μg/ml多肽;
46.所述多肽的氨基酸序列如seq id no.1所示。
47.优选地,所述多肽的浓度为5μg/ml。
48.与现有技术相比,本发明具有如下的有益效果:
49.1、本发明从omd核心蛋白中筛选得到了其11段lrrs基序的第10-11段的多肽,并证明了该多肽能与bmp2结合,并与bmp受体形成复合物,从而激活下游smads调节转录反应,减少bmp2用量,优化bmp2成骨。
50.2、本发明筛选得到的多肽分子量小,结构简单且稳定,而且活性更高(omd可促进bmp2诱导的sp7表达约10倍,而本发明的多肽可提高至50倍以上),可更好的应用于促进骨损伤再生药物中。
附图说明
51.通过阅读参照以下附图对非限制性实施例所作的详细描述,本发明的其它特征、目的和优点将会变得更明显:
52.图1为本发明制备的不同lrr多肽对bmp2诱导的人牙髓干细胞(hdpscs)的碱性磷酸酶活性检测结果;
53.图2为本发明制备的lrr10-11多肽的热变性测定结果图;
54.图3为本发明制备的lrr10-11多肽对hdpscs的毒性测试结果;
55.图4为本发明制备的lrr10-11多肽在不同浓度下对bmp2诱导的hdpscs的碱性磷酸酶活性的结果;
56.图5为本发明制备的lrr10-11多肽对bmp2诱导的hdpscs的成骨相关基因表达的结
果;
57.图6为本发明制备的lrr10-11多肽对bmp/smad信号通路的作用结果。
具体实施方式
58.下面结合具体实施例对本发明进行详细说明。以下实施例将有助于本领域的技术人员进一步理解本发明,但不以任何形式限制本发明。应当指出的是,对本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进。这些都属于本发明的保护范围。
59.实施例1
60.本实施例提供了一种促进bmp2诱导成骨功能的多肽的制备方法,包括以下步骤:
61.1.构建过表达质粒
62.1.1获得引物
63.以pgex-4t-1为载体,选取bamhi和noti为酶切位点,设计omd中不同lrr片段:omd_lrrnt,omd_lrr1-3,omd_lrr4-6,omd_lrr7-9,omd_lrr10-11的引物。
64.omd_lrrnt的氨基酸序列(seqidno:2)为:vpfhqytlgcvsecfcptnfpssmycdnrklktipnipm;
65.omd_lrr1-3的氨基酸序列(seqidno:3)为:hiqqlylqfneieavtansfinathlkeinlshnkiksqkidygvfaklpnllqlhlehnnleefpfplpksl;
66.omd_lrr4-6的氨基酸序列(seqidno:4)为:erlllgyneisklqtnamdglvnltmldlcynylhdsllkdkifakmeklmqlnlcsnrlesmppglps;
67.omd_lrr7-9的氨基酸序列(seqidno:5)为:slmylslennsissipekyfdklpklhtlrmshnklqdipynifnlpnivelsvghnklkq;
68.omd_lrr10-11的氨基酸序列(seqidno:1)为:nlehlylqnneiekmnltvmcpsidplhyhhltyirvdqnklkepissyiffc。
69.各引物的获得是通过在pubmedgene中找到各lrr片段的编码产物mrna的cds,设计补上终止密码子,得到各lrr片段的引物序列如下表1所示:
70.表1
[0071][0072]
1.2获得目的基因
[0073]
使用pcr获得各目的基因片段,pcr反应体系如表2所示:
[0074]
表2
[0075]
试剂体积终浓度primestar max premix(2
×
)25μl1
×
primer forward1.5μl0.3μmprimer reverse1.5μl0.3μmtemplate1μl ddh2o21μl [0076]
采用的质粒dna为2ng。
[0077]
pcr扩增条件为表3所示的温度和时间,35个循环。
[0078]
表3
[0079][0080]
[0081]
1.3跑胶鉴定
[0082]
①
3%琼脂糖凝胶:1.5g琼脂+50ml tae buffer加热2min,等待冷却至60-70℃,加入5μl 4s green nucleic acid stain。
[0083]
②
用10
×
loading buffer稀释步骤1.2获得的pcr产物,上dl500 dna marker,设置电泳槽130v 30min。
[0084]
1.4使用tiangel midi purification kit(tiangen)进行dna胶回收以获得纯净的各 lrr dna片段。
[0085]
1.5双酶切pgex-4t-1载体,获得线性质粒
[0086]
采用表4所示的双酶切反应体系,30℃加热5min,37℃加热5min,得到线性质粒。
[0087]
表4
[0088]
试剂体积10
×
quickcut green buffer2μlplasmid dna(pgex-4t-1)1μgquickcut
tm
bamh i1μlquickcut
tm
not i1μlddh2oup to 20μl
[0089]
1.6使用in-hd cloning kit(takara)将步骤1.4获得的各目的基因片段(即 lrr dna片段)插入到双酶切后的pgex-4t-1载体中,获得重组质粒。
[0090]
1.7将步骤1.6获得的重组质粒转化top10大肠杆菌,挑细菌单克隆小摇,使用 tianprep mini plasmid kit(tiangen)进行质粒小量抽提,酶切鉴定,送公司测序鉴定。鉴定正确质粒转化top10大肠杆菌,然后挑细菌单克隆大摇,使用endofree maxiplasmid kit(tiangen)进行质粒大量抽提纯化备用,即得到过表达质粒。
[0091]
2.使用大肠杆菌bl21获得各lrr多肽
[0092]
2.1取步骤1获得的测序正确的过表达质粒,转化bl21感受态;
[0093]
2.2取10ml lb培养基,含有1:2000(体积比)氨苄青霉素(氨苄青霉素母液: 100mg/ml),挑步骤2.1获得的细菌单克隆,37℃摇床培养6-7h后,将摇得的菌液以 1:20体积比转接入含200ml lb培养基的锥形瓶中,37℃摇床培养1.5小时后测od
600
数值,待od
600
值达到0.6后,加入100μm iptg 20℃处理12-14小时,诱导蛋白表达。
[0094]
2.3将步骤2.2处理得到的bl21菌液在5000g、4℃下离心5min,然后使用0.2 mg/ml溶菌酶、20μg/ml dnase、1mm mgcl2、1mm pmsf在4℃下裂解30min。再将裂解的液体转移到离心管中在4℃、12000rmp下离心20min,然后用0.45μm的滤膜过滤。
[0095]
2.4使用gst-sefinose(tm)kit(sangon biotech)试剂盒提取多肽。
[0096]
①
用10倍树脂体积的binding/wash buffer平衡柱子,流速为0.5~1ml/min,使缓冲液完全流出或a280达到基线。
[0097]
②
将步骤2.3过滤后的裂解液加入到树脂平衡柱并将树脂与液体混匀,流速为 0.5~1ml/min。如有需要,可将流穿液再次加入到树脂中直至树脂达到最大载量。
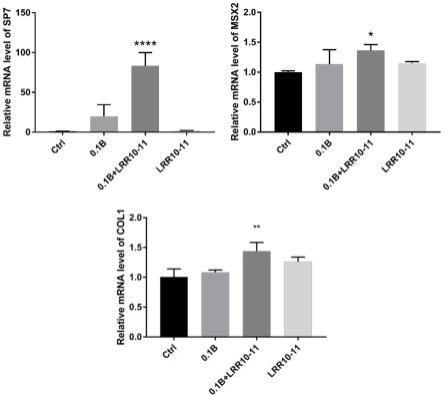
[0098]
③
加入5~10倍柱体积的binding/wash buffer冲洗树脂。重复清洗步骤,通过测量在 280nm吸光度,直到洗出液值达到基线值。
[0099]
④
用2倍柱体积的elution buffer洗脱gst标签蛋白。通过测量在280nm处的馏分
的吸光度监测蛋白洗脱情况。
[0100]
3.使用高容量内毒素去除树脂(pierce high-capacity endotoxin removal resin)去除内毒素后获得各多肽。
[0101]
3.1将树脂柱恢复至室温;
[0102]
3.2拧开树脂柱底部封口,松开顶盖。将树脂柱放入收集管中。将柱子以500
×
g的速度离心1min,弃去储存液。
[0103]
3.3拆下柱盖并插入底部塞子。加入0.2n naoh,盖紧盖子,将柱子倒置几次,直到树脂悬浮在溶液中并在室温下孵化过夜。
[0104]
3.4松开盖子,拆下底部封口。将树脂柱放入收集管中。将柱子以500
×
g的速度离心1min,弃去储存液。
[0105]
3.5拆下柱盖并插入底部塞子。加入2m nacl,盖紧盖子,将柱子倒置几次,直到树脂悬浮在溶液中。
[0106]
3.6重复步骤3.4。
[0107]
3.7拆下柱盖并插入底部塞子。加入不含内毒素的超纯水。盖紧盖子,将柱子倒置几次,直到树脂悬浮在溶液中。
[0108]
3.8重复步骤3.4。
[0109]
3.9拆下柱盖并插入底部塞子。加入无内毒素的缓冲液。盖紧盖子,将柱子倒置几次,直到树脂悬浮在溶液中。
[0110]
3.10重复步骤3.4。再重复步骤3.9和3.10两次。
[0111]
3.11拆下柱盖并插入底部塞子。加入步骤2处理后得到的样品。盖紧盖子,将柱子倒置几次,直到树脂悬浮在溶液中。
[0112]
3.12在室温下,360
°
摇床温和混匀1小时。
[0113]
3.13松开盖子,拆下底部封口。将树脂柱放入收集管中。将柱子以500
×
g的速度离心1min,收集样品。
[0114]
实施例2碱性磷酸酶活性检测
[0115]
碱性磷酸酶(alp)是成熟成骨细胞的标志性酶,也是成骨细胞分化的标志之一,可增加局部无机磷酸的浓度,参与钙化组织的形成、代谢和再生。pnpp是一种常用的磷酸酶显色底物,在碱性条件下,可在碱性磷酸酶作用下生成para-nitrophenol,呈黄色产物,可以在400-415nm检测吸光度。产物黄色越深,说明碱性磷酸酶检活性越高。
[0116]
本实施例进行碱性磷酸酶(alp)活性检测的步骤为:
[0117]
(1)将hdpscs接种到12孔板中,待细胞长至90%密度时加下述分组的蛋白刺激,分组为:ctrl组(为0.1%bsa溶液),0.1b组(为含0.1μg/ml bmp2的0.1%bsa溶液),0.1b+1o_组(为含0.1μg/ml bmp2+1μg/ml omd的0.1%bsa溶液),0.1b+3o 组(为含0.1μg/ml bmp2+3μg/ml omd的0.1%bsa溶液),0.1b+1lrrnt组(为含 0.1μg/ml bmp2+1μg/ml omd_lrrnt的0.1%bsa溶液),0.1b+1lrr1-3组(为含 0.1μg/ml bmp2+1μg/ml omd_lrr1-3的0.1%bsa溶液),0.1b+1lrr4-6组(为含 0.1μg/ml bmp2+1μg/ml omd_lrr4-6的0.1%bsa溶液),0.1b+1lrr7-9组(为含 0.1μg/ml bmp2+1μg/ml omd_lrr7-9的0.1%bsa溶液),0.1b+1lrr10-11组(为含0.1μg/ml bmp2+1μg/ml omd_lrr10-11的0.1%bsa溶液),0.1b+1gst组(为含0.1μg/ml bmp2+1μg/ml gst的0.1%bsa溶液);每2天换液一次;
[0118]
(2)准备样品:4天后取出培养皿,用pbs洗涤1次,吸干,用适量lysis buffer 裂解细胞,离心取上清作为样品;
[0119]
(3)准备空白对照和标准品:准备96孔板,每个孔应有3个复孔,在样品孔中加入显色底物50μl+样品50μl,混匀;
[0120]
(4)37℃孵育5-10min,可延长至30min;
[0121]
(5)每孔加入100μl反应终止液终止反应,在405nm测定吸光度。
[0122]
结果如图1所示,0.1b+3o组和0.1b+1lrr10-11组可显著促进bmp2诱导的alp 活性,且0.1b+1lrr10-11组促进bmp2诱导的alp活性明显高于0.1b+3o组,但其采用的omd_lrr10-11多肽的浓度却仅为omd浓度的1/3。
[0123]
实施例3 omd_lrr10-11热变性测定
[0124]
蛋白去折叠转变温度,tm,是指有50%蛋白质去折叠时的温度。高tm的蛋白质更稳定,因为达到转变温度时需要更大的能量输入。tm是一种准确且公认的的衡量蛋白质稳定性的指标,所以它是一个必须确定的参数。本实施例用于测量多肽 omd_lrr10-11的蛋白质稳定性。
[0125]
本实验采用的仪器为prometheus(pr)(nanotemper,德国)。
[0126]
实验步骤为:用毛细玻璃管吸取一定浓度实施例1方法获得的多肽溶液(无气泡),然后使用prometheus(pr)测定tm。通过检测蛋白的内源荧光变化来观察蛋白的折叠状态。将单波长(350或330nm)的荧光强度或350/330的荧光强度比值,与温度的升高或化学变性剂的浓度进行拟合,以确定蛋白质的tm。结果如图2所示,多肽 omd_lrr10-11的tm为52.2℃,证明其具有较好的稳定性。
[0127]
实施例4 omd_lrr10-11细胞毒性测定
[0128]
cck8试剂可用于细胞毒性分析,其基本原理为:试剂中含有wst
–
8:它在电子载体1-methoxy pms的作用下被细胞线粒体中的脱氢酶还原为具有高度水溶性的黄色甲臜产物,生成的甲臜物的数量与活细胞的数量成正比。应用此特性进行细胞毒性试验分析等。
[0129]
本实施例进行细胞毒性实验分析的步骤为:
[0130]
(1)将hdpscs悬浮液计数并以2
×
103/孔的密度接种到96孔板中,每孔加 100μl 生长培养基;
[0131]
(2)每个96孔板设置1个对照组(omd_lrr10-11浓度为0μg/ml的0.1%bsa 溶液)和3个实验组(omd_lrr10-11浓度分别为0.2μg/ml、2μg/ml、20μg/ml的0.1%bsa溶液),每组设3个复孔。4个组从高到低加入不同浓度的omd_lrr10-11 溶液(0-20μg/ml)培养24h;
[0132]
(3)至预设检测时间点时,将cck8溶液与双无dmem培养基按1:9混合形成cck8稀释液,每孔加100μl cck8稀释液,避光,于恒温培养箱中孵育1h;
[0133]
(4)酶标仪检测吸光度:利用酶标仪,检测波长为450nm时的吸光度,分析数据。
[0134]
结果如图3所示,omd_lrr10-11在0.2-20μg/ml范围内对细胞的活力没有影响。
[0135]
实施例5不同浓度的omd_lrr10-11的碱性磷酸酶活性检测
[0136]
碱性磷酸酶(alp)是成熟成骨细胞的标志性酶,也是成骨细胞分化的标志之一,可增加局部无机磷酸的浓度,参与钙化组织的形成、代谢和再生。pnpp是一种常用的磷酸酶显色底物,在碱性条件下,可在碱性磷酸酶作用下生成para-nitrophenol,呈黄色产物,可以在400-415nm检测吸光度。产物黄色越深,说明碱性磷酸酶检活性越高。
[0137]
本实施例进行alp活性检测的步骤为:
[0138]
(1)将hdpscs接种到12孔板中,待细胞长至90%密度时加下述分组的蛋白刺激,分组为:ctrl组(为0.1%bsa溶液),0.1b组(为含0.1μg/mlbmp2的0.1%bsa溶液),0.1b+0.02lrr10-11组(为含0.1μg/mlbmp2+0.02μg/mlomd_lrr10-11的0.1%bsa溶液),0.1b+0.2lrr10-11组(为含0.1μg/mlbmp2+0.2μg/mlomd_lrr10-11的0.1%bsa溶液),0.1b+2lrr10-11组(为含0.1μg/mlbmp2+2μg/mlomd_lrr10-11的0.1%bsa溶液),0.1b+20lrr10-11组(为含0.1μg/mlbmp2+20μg/mlomd_lrr10-11的0.1%bsa溶液);每2天换液一次;
[0139]
(2)准备样品:4天后取出培养皿,用pbs洗涤1次,吸干,用适量lysisbuffer裂解细胞,离心取上清作为样品;
[0140]
(3)准备空白对照和标准品:准备96孔板,每个孔应有3个复孔,在样品孔中加入显色底物50μl+样品50μl,混匀;
[0141]
(4)37℃孵育5-10min,可延长至30min;
[0142]
(5)每孔加入100μl反应终止液终止反应,在405nm测定吸光度。
[0143]
结果如图4所示,omd_lrr10-11在浓度为0.02-20μg/ml下均可促进bmp2诱导的alp活性。尤其是0.1b+0.2lrr10-11组和0.1b+2lrr10-11组的alp活性显著高于其他组。
[0144]
实施例6omd_lrr10-11对成骨相关基因表达的作用
[0145]
sp7是一种含锌指的成骨细胞特异性转录因子,在成骨细胞分化和骨矿化过程起到了重要的作用。bmp2/smads/msx2/sp7信号通路是骨组织形成过程中mscs分化为成骨细胞及骨细胞外基质合成分泌所必需的。骨含有结构大分子和其他蛋白质作为细胞外基质成分,其中最主要的是i型胶原(col1)。
[0146]
本实施例进行成骨相关基因表达的步骤为:
[0147]
1)本实施例进行的分组为:对照组(ctrl)为采用0.1%bsa溶液,实验组1(0.1b)为采用含0.1μg/mlbmp2的0.1%bsa溶液,实验组2(0.1b+lrr10-11)为采用含0.1μg/mlbmp2和5μg/mlomd_lrr10-11的0.1%bsa溶液,实验组4(lrr10-11)为采用含5μg/mlomd_lrr10-11的0.1%bsa溶液。
[0148]
2)将hdpscs接种到12孔板中,待细胞长至90%密度时加上述分组的蛋白刺激,分别24h和48h收样。
[0149]
3)按生产说明书用trizol试剂提取人dpscs总rna。分离的rna在分光光度计上定量。用primescriptrt试剂盒(takara,kusatsu,japan)将260/280比值~2.0的500ngrna逆转录成互补dna(cdna)。在lightcycler480ii仪器上使用tbgreenpremixextaq(takara)测定基因转录水平。反应程序为40个循环,95℃变性5s,55℃退火30s,72℃延伸30s,重复三次。以人actin为内参,并运用2
‑△△
ct值计算各基因的相对表达量。各基因的引物序列如下表5所示:
[0150]
表5
[0151][0152]
各基因的表达结果如图5所示,0.1b+lrr10-11组相比其他组可明显提高col1、 msx2、sp7的表达,且单独使用lrr10-11不能提高col1和msx2的表达。
[0153]
实施例7 omd_lrr10-11对bmp/smads信号通路影响
[0154]
bmp2是目前已知的生长因子中诱导成骨细胞分化和骨形成作用强而有效的一种,是涉及成骨信号事件并最终产生功能性骨组织的生物级联反应的发起者。 bmp2与跨膜型丝氨酸-苏氨酸激酶受体络合传递信号,受体分为i型和ii型两个亚型,在配体结合后,ii型受体使i型受体磷酸化。激活的i型受体招募并磷酸化通路特异性r-smads(smad1、smad5和smad8等),与smad4形成三聚体并转运到细胞核,与dna结合,通过招募染色质重塑机制和与组织特异性转录因子整合来调节基因表达。
[0155]
本实施例进行实验的步骤为:
[0156]
1)实验分组:对照组(bmp2和lrr10-11的浓度均为0μg/ml的0.1%bsa溶液),实验组1(bmp2的浓度为0.02μg/ml的0.1%bsa溶液),实验组2(bmp2的浓度为 0.02μg/ml、lrr10-11的浓度为5μg/ml的0.1%bsa溶液)。
[0157]
2)将hdpscs接种到6孔板中,待细胞长至90%密度时无血清dmem饥饿24h后加上述分组的蛋白刺激,30min收样。
[0158]
3)用含有1mm pmsf的细胞裂解缓冲液提取蛋白,用bradford蛋白分析试剂盒 (beyotime)定量。用10%sds聚丙烯酰胺凝胶分离等量的蛋白质,然后转移到pvdf膜上。5%牛奶封闭液封闭pvdf膜,一抗4℃温和摇晃过夜。二抗室温下孵育目的条带 1h后,显影液检测条带,并用化学发光成像系统显示条带。
[0159]
结果如图6所示,omd_lrr10-11可激活bmp/smads信号通路。
[0160]
本发明具体应用途径很多,以上所述仅是本发明的优选实施方式。应当指出,以上实施例仅用于说明本发明,而并不用于限制本发明的保护范围。对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进,这些改进也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1