一种外消旋米格列奈的制备方法与流程
1.本发明涉及有机化学技术领域,尤其涉及一种外消旋米格列奈的制备方法。
背景技术:
2.米格列奈(mitiglinide),化学式为(2s)-2-苄基-3-(顺式全氢异吲哚-2-羰基)丙酸,分子式c
19h25
no3,米格列奈的钙盐(mitiglinide calcium dihydrate)可用于治疗ii型糖尿病,于2004年首次在日本上市。其作用机理是米格列奈与胰岛β细胞膜上磺酰脲受体结合,关闭β细胞膜上atp依赖性k
+
通道,导致ca
2+
内流,细胞内ca
2+
浓度升高,从而促进胰岛素分泌,降低血糖。对于米格列奈钙的制备,近年来已有较多合成专利报道。专利cn10210838a、cn102898348a等对米格列奈钙的合成路线进行了详细的报道,其中关键中间体(s)-2-苄基琥珀酸的制备条件较为苛刻,常用到pd/c催化剂加氢还原,同时合成步骤后处理繁琐,容易造成原料的浪费,增加了工业生产的成本,不符合“碳中和”发展理念。
3.
技术实现要素:
4.本发明要解决的技术问题是克服现有技术存在的缺陷,本发明提出了一种外消旋米格列奈的制备方法,成本较低,选择性高,反应条件温和,后处理简便,能以较高产率合成外消旋米格列奈,以解决现有技术中的问题。
5.为解决上述技术问题,本发明采用的技术方案是:一种外消旋米格列奈的制备方法,采用分步反应,包括如下步骤:
6.s1、3-氰基-4苯基-n-(8-氨基喹啉)-3-丁酰胺的合成:以n-(8-氨基喹啉)-3-丁烯酰胺为原料,以芳氰化合成方式纯化后制得3-氰基-4苯基-n-(8-氨基喹啉)-3-丁酰胺;
7.s2、2-苄基琥珀酸的合成:置3-氰基-4苯基-n-(8-氨基喹啉)-3-丁酰胺、氢氧化钾、乙醇和水于反应瓶中回流反应36h,反应结束后以乙酸乙酯多次萃取所得反应液并合并有机相,经纯化浓缩干燥后即得2-苄基琥珀酸;
8.s3、2-苄基丁二酸酐的合成:于惰性气氛下,2-苄基琥珀酸与三氟乙酸酐于0℃下完成反应,除去所得反应液中易挥发组分即得2-苄基丁二酸酐;
9.s4、外消旋米格列奈的合成:溶解2苄基丁二酸酐于溶剂中,于0℃下向溶液中滴入顺式全氢异吲哚完成反应,反应液经纯化洗涤干燥之后即得外消旋米格列奈。
10.进一步地,所述芳氰化合成过程中,原料包括n-(8-氨基喹啉)-3-丁烯酰胺、碘苯、氰基甲酸乙酯、双(三氟甲磺酸)镍以及磷酸钠,所述原料摩尔比为1:2-3:1.5-2:0.1-0.15:1.5-3;
11.所用反应溶剂为1,4-二氧六环、四氢呋喃、甲苯、乙腈、乙二醇二甲醚中的任意一种。
12.进一步地,所述芳氰化合成过程于惰性气氛下在100-130℃搅拌反应24-36h,于反应结束后将混合物减压蒸发去除溶剂得到粗产物,将粗产物经过色谱柱纯化,得产物3-氰基-4苯基-n-(8-氨基喹啉)-3-丁酰胺。
13.进一步地,所述色谱柱填充物为硅胶,粗产物经色谱柱洗脱,洗脱液为乙酸乙酯和石油醚的混合物,乙酸乙酯和石油醚的体积比为1:3-5。
14.进一步地,于s2中,3-氰基-4-苯基-n-(8-氨基喹啉)-3-丁酰胺与氢氧化钾的摩尔比为1:5-20。
15.进一步地,回流反应于90-120℃下进行,反应结束后以乙酸乙酯萃取三次去除8-氨基喹啉后以盐酸调节水相ph至1-3,随后以乙酸乙酯萃取水相得2-苄基琥珀酸;所述盐酸浓度为3m。
16.进一步地,于s3中,所述反应过程于0℃下反应2-4h,除去反应液中易挥发组分过程于真空环境下进行。
17.进一步地,于s4中,2苄基丁二酸酐与顺式全氢异吲哚摩尔比为1:1-1.5;所述溶剂为二氯甲烷或甲苯中任意一种。
18.进一步地,反应液于25-50℃反应2-5h,于反应结束后减压蒸发出去二氯甲烷后以异丙醚洗涤,所得产物于真空环境下进行干燥即得外消旋米格列奈。
19.进一步地,所述n-(8-氨基喹啉)-3-丁烯酰胺的合成过程具体如下:
20.于反应容器中依次加入足量乙烯基乙酸、8-氨基喹啉、吡啶、2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu)和二氯甲烷,反应于室温下进行直至8-氨基喹啉完全转化,向所述反应液中加入饱和碳酸氢钠溶液搅拌;
21.所得混合液以二氯甲烷萃取,合并有机相并以饱和食盐水洗涤,于真空环境下浓缩后剩余物通过柱色谱分离纯化得到n-(8-氨基喹啉)-3-丁烯酰胺。
22.与现有技术相比,本发明的有益效果包括:整个过程从乙烯基乙酸开始,经过5步以50%的总收率得到外消旋的米格列奈;各步骤反应条件温和,后处理纯化过程简单,且每步产物收率可达80%以上,最高可达90%,产率高,选择性好,原料损失少,经济环保等优点,可有效减少原料的浪费,进而可节约生产成本,适用于工业化生产。
附图说明
23.参照附图来说明本发明的公开内容。应当了解,附图仅仅用于说明目的,而并非意在对本发明的保护范围构成限制。在附图中,相同的附图标记用于指代相同的部件。其中:
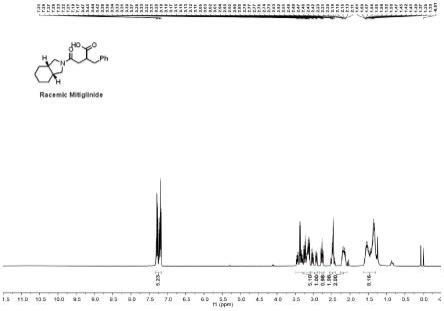
24.图1示为外消旋米格列奈的核磁共振氢谱图。
具体实施方式
25.容易理解,根据本发明的技术方案,在不变更本发明实质精神下,本领域的一般技术人员可以提出可相互替换的多种结构方式以及实现方式。因此,以下具体实施方式以及附图仅是对本发明的技术方案的示例性说明,而不应当视为本发明的全部或者视为对本发明技术方案的限定或限制。
26.一种外消旋的米格列奈的制备方法,合成路径如下:
[0027][0028]
包括以下步骤:
[0029]
步骤一、n-(8-氨基喹啉)-3-丁烯酰胺的合成
[0030]
在反应容器中,依次加入乙烯基乙酸、8-氨基喹啉、吡啶、2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu)和二氯甲烷,反应在室温下搅拌过夜,tlc板监测。8-氨基喹啉完全转化后,加入饱和nahco3溶液搅拌。混合物用二氯甲烷萃取三遍,有机相用饱和食盐水洗涤,在真空下浓缩后剩余物通过柱色谱分离纯化得到n-(8-氨基喹啉)-3-丁烯酰胺。
[0031]
步骤二、3-氰基-4苯基-n-(8-氨基喹啉)-3-丁酰胺的合成
[0032]
在惰性气体条件下,向反应瓶中加入n-(8-氨基喹啉)-3-丁烯酰胺、碘苯、氰基甲酸乙酯、双(三氟甲磺酸)镍、磷酸钠和150ml 1,4-二氧六环,反应在130℃下搅拌24小时。反应结束后混合物冷却到室温,过滤,滤液经真空浓缩后剩余物通过柱色谱分离纯化得到3-氰基-4苯基-n-(8-氨基喹啉)-3-丁酰胺。
[0033]
步骤三、2-苄基琥珀酸的合成
[0034]
在反应瓶中,加入3-氰基-4苯基-n-(8-氨基喹啉)-3-丁酰胺、氢氧化钾、乙醇和水,回流反应36小时。反应结束后将反应液冷却至室温,用乙酸乙酯萃取三次除去8-氨基喹啉,剩余水相用盐酸溶液调节ph至1-3,用乙酸乙酯萃取三次,所得有机相用饱和食盐水洗涤,在真空下浓缩干燥得2-苄基琥珀酸。
[0035]
步骤四、2-苄基丁二酸酐的合成
[0036]
在惰性气体条件下,于0℃下向反应瓶中加入2-苄基琥珀酸和三氟乙酸酐,随后反应液在室温下搅拌。反应液在真空下除去易挥发组分,得到2-苄基丁二酸酐。
[0037]
步骤五、外消旋米格列奈的合成
[0038]
将步骤四所得2-苄基丁二酸酐溶于二氯甲烷中,在0℃下向该溶液逐滴加入顺式全氢异吲哚,反应液在室温下搅拌。反应完成后用旋转蒸发仪除去二氯甲烷,剩余物用异丙醚洗涤,在真空下干燥,得到黄色粘稠状的外消旋米格列奈。
[0039]
以下结合实施例对于本技术的技术效果作进一步说明。
[0040]
实施例1
[0041]
(1)在250ml圆底烧瓶中,依次加入24mmol乙烯基乙酸、20mmol 8-氨基喹啉、40mmol吡啶、26mmol 2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu)和50ml二氯甲烷,反应在室温下搅拌过夜,tlc板监测。8-氨基喹啉完全转化后,加入饱和
nahco3溶液搅拌30分钟。混合物用二氯甲烷萃取三遍,有机相用饱和食盐水洗涤,在真空下浓缩后剩余物通过柱色谱分离纯化得到n-(8-氨基喹啉)-3-丁烯酰胺,称重3.82g,收率为90%。
[0042]
(2)在氩气条件下,向250m史莱克瓶中加入10mmol n-(8-氨基喹啉)-3-丁烯酰胺、30mmol碘苯、20mmol氰基甲酸乙酯、1mmol双(三氟甲磺酸)镍、20mmol磷酸钠和150ml无水1,4-二氧六环,反应在130℃下搅拌24小时。反应结束后混合物冷却到室温,过滤,滤液经真空浓缩后剩余物通过柱色谱分离纯化得到3-氰基-4苯基-n-(8-氨基喹啉)-3-丁酰胺,称重2.52g,收率为80%。
[0043]
(3)在50ml圆底烧瓶中,加入5mmol 3-氰基-4苯基-n-(8-氨基喹啉)-3-丁酰胺、100mmol氢氧化钾、15ml乙醇和15ml水,反应在100℃下搅拌36小时。反应结束后将反应液冷却至室温,用乙酸乙酯萃取三次除去8-氨基喹啉,剩余水相用3m盐酸溶液调节ph至2-3,用乙酸乙酯萃取三次,所得有机相用饱和食盐水洗涤,在真空下浓缩干燥得2-苄基琥珀酸,称重0.83g,收率为80%。
[0044]
(4)在氩气条件下,于0℃下向25ml史莱克瓶中加入3mmol 2-苄基琥珀酸和3.3mmol三氟乙酸酐,随后反应液在室温下搅拌2小时。反应液在真空下除去易挥发组分,得到2-苄基丁二酸酐,称重0.52g,收率为90%。
[0045]
(5)将步骤四所得2-苄基丁二酸酐溶于20ml无水二氯甲烷中,在0℃下向该溶液逐滴加入5mmol顺式全氢异吲哚,反应液在室温下搅拌3小时。反应完成后用旋转蒸发仪除去二氯甲烷,剩余物用异丙醚洗涤,在真空下干燥,得到黄色粘稠状的外消旋米格列奈,称重0.77g,收率为90%,其核磁共振氢谱图如图1所示。
[0046]
本发明的技术范围不仅仅局限于上述说明中的内容,本领域技术人员可以在不脱离本发明技术思想的前提下,对上述实施例进行多种变形和修改,而这些变形和修改均应当属于本发明的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1