一对具有抗炎活性的聚酮类化合物及其制备方法和应用
1.本发明属于医药技术领域,具体涉及一对具有抗炎活性的新骨架聚酮类差向异构体及其制备方法和应用。
背景技术:
2.药用植物内生菌具有多样性特征,每个内生菌又有丰富的次级代谢产物,目前已从内生菌次级代谢产物中发现了许多具有生物活性的生物碱、多肽、聚酮类、萜类等物质,这些次级代谢物不但具有与宿主植物相同或相似的生物活性,例如抗菌、抗炎、抗肿瘤、抗高血糖、抗寄生虫等,还具有许多新的生物活性。因此药用植物内生菌的次级代谢产物是庞大的潜在药用物质资源库,具有良好的开发前景。
技术实现要素:
3.鉴于此,本发明的目的是提供了一对具有抗炎活性的聚酮类化合物及其制备方法和应用,本发明通过对植物内生菌进行研究,充分的利用植物内生菌资源,从采集自西藏红景天rhodiola tibetica的叶片部位分离获得链格孢属内生真菌(alternaria sp.)hjt-y7,于2022年5月6日保藏于中国微生物菌种保藏管理委员会普通微生物中心,保藏编号为cgmcc no.40166,保藏单位地址为北京市朝阳区北辰西路一号院三号,对获得的链格孢属内生真菌(alternaria sp.)hjt-y7进行固体发酵,使用一系列分离纯化方法分离获得一对结构新颖的聚酮类差向异构体,为药物开发提供了新的先导化合物。
4.本发明目的是通过以下方式实现:
5.本发明提供一对聚酮类化合物或其药学上可接受的盐,所述的聚酮类化合物的结构如下所示:
[0006][0007]
本发明另一方面提供一株链格孢属内生真菌(alternaria sp.)hjt-y7,于2022年5月6日保藏于中国微生物菌种保藏管理委员会普通微生物中心,保藏编号为cgmcc no.40166,保藏单位地址为北京市朝阳区北辰西路一号院三号。
[0008]
本发明另一方面提供所述的聚酮类化合物的制备方法,主要包括如下步骤:
[0009]
(1)将上述的链格孢属内生真菌(alternaria sp.)hjt-y7接种到真菌4号培养基中进行震荡培养,将所得的发酵液连同菌丝体接种于大米固体培养基中,20~30℃下静止发酵10~40天,得到固体发酵产物;
[0010]
(2)将步骤(1)得到的固体发酵产物使用甲醇进行超声提取,过滤,所得的提取液上样至硅胶色谱柱,梯度洗脱,流动相为氯仿/甲醇,氯仿/甲醇的体积比由100:0梯度降低至0:100,根据tlc结果,依次得到组份1-16;将组份7上样至硅胶色谱柱,梯度洗脱,流动相为石油醚/乙酸乙酯,石油醚/乙酸乙酯的体积比由10:1梯度降低至10:9,根据tlc结果,依次得到组份7-1~7-10;将组份7-6上样至sephadex lh-20凝胶柱,流动相为二氯甲烷/甲醇体积比为1:1的混合液,依次得到组份7-6-1~7-6-6,将组份7-6-3上样至agilent c18高效液相色谱,流动相为55%甲醇水溶液,得到化合物1和化合物2。
[0011]
进一步的,步骤(1)中所述的震荡培养的条件为20~30℃,100~200r/min。
[0012]
进一步的,步骤(1)中所述的大米固体培养基是由大米和纯净水按质量体积比为80:80~120g/ml配制而成。
[0013]
进一步地,步骤(1)中所述的真菌4号培养基组成为:2%甘露醇,2%葡萄糖,0.5%酵母膏,1%蛋白胨0.05%kh2po4,0.03%mgso4·
7h2o,0.1%玉米浆,去离子水。
[0014]
进一步地,步骤(2)中所述梯度洗脱过程中氯仿/甲醇的体积比具体为100:0、100:1、100:2、100:3、100:5、100:10、0:100。
[0015]
进一步地,步骤(2)中所述梯度洗脱过程中石油醚/乙酸乙酯的体积比具体为10:1、10:3、10:5、10:6、10:7、10:9。
[0016]
本发明另一方面提供了一种药物组合物,其包含所述的聚酮类化合物或其药学上可接受的盐与药学上可接受的辅料。
[0017]
本发明还提供了所述聚酮类化合物或其药学上可接受的盐或所述的药物组合物在制备抗炎药物中的应用。
[0018]
本发明相对于现有技术具有的有益效果如下:
[0019]
本发明充分利用植物内生菌资源,对植物内生菌进行研究,寻找新的聚酮类活性化合物,并对其进行抗炎活性实验,与模型组相比,化合物1和化合物2在各浓度下均对no的释放有不同程度的抑制,在5μm的浓度下均表现出比阳性对照组更优的抑制能力,表明化合物1与化合物2具有优良的抗炎效果。
附图说明
[0020]
为了更清楚地说明本发明实施例,下面将对实施例涉及的附图进行简单地介绍。
[0021]
图1为实施例1分离的化合物1的1h-nmr图;
[0022]
图2为实施例1分离的化合物1的
13
c-nmr图;
[0023]
图3为实施例1分离的化合物1的hsqc图;
[0024]
图4为实施例1分离的化合物1的hmbc图;
[0025]
图5为实施例1分离的化合物1的noesy图;
[0026]
图6为实施例1分离的化合物2的1h-nmr图;
[0027]
图7为实施例1分离的化合物2的
13
c-nmr图;
[0028]
图8为实施例1分离的化合物2的hsqc图;
[0029]
图9为实施例1分离的化合物2的hmbc图;
[0030]
图10为实施例1分离的化合物2的noesy图;
[0031]
图11为实施例1分离的化合物1-2的cd图;
[0032]
图12为实施例1分离的化合物1-2的dp4+图;
[0033]
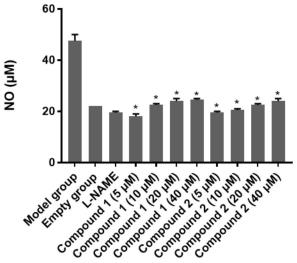
图13为实施例1分离的化合物1-2的抗炎效果图。
具体实施方式
[0034]
下面结合实施例对本发明进行详细的说明,但本发明的实施方式不限于此,显而易见地,下面描述中的实施例仅是本发明的部分实施例,对于本领域技术人员来讲,在不付出创造性劳动性的前提下,获得其他的类似的实施例均落入本发明的保护范围。如无特殊说明,本发明所采用的实验方法均为常规方法,所用实验器材、材料、试剂等均可从商业途径获得。
[0035]
实施例1
[0036]
一对聚酮类化合物的制备方法,主要包括如下步骤:
[0037]
(1)菌种发酵:将采集自西藏红景天叶片部位的链格孢属内生真菌(alternaria sp.)hjt-y7采用真菌4号培养基(2%甘露醇,2%葡萄糖,0.5%酵母膏,1%蛋白胨0.05%kh2po4,0.03%mgso4·
7h2o,0.1%玉米浆,去离子水配制)进行震荡培养,培养条件28℃,180r/min,再将震荡培养的发酵液连同菌丝体接种于装有大米固体培养基(大米和纯净水按质量体积比为80g:110ml配制)的锥形瓶中静止发酵培养40天,培养温度为28℃,得到固体发酵产物;
[0038]
(2)代谢产物的提取分离:将步骤(1)得到的固体发酵产物使用等体积甲醇进行超声提取,经8层纱布过滤,将提取液与菌丝体及大米分离,浓缩提取液,提取液上样至硅胶色谱柱进行分离,以300-400目硅胶为固定相,以氯仿/甲醇为流动相进行梯度洗脱,梯度依次为氯仿:甲醇=100:0、100:1、100:2、100:3、100:5、100:10、0:100(v/v),洗脱剂流速为30ml/min,每个梯度洗脱10000ml,收集洗脱液(每500ml收集一瓶),将洗脱液浓缩,根据tlc结果合瓶,依次得到1-16组份;将组份7上样至硅胶色谱柱,以200-300目硅胶为固定相,流动相为石油醚/乙酸乙酯,梯度依次为石油醚:乙酸乙酯=10:1、10:3、10:5、10:6、10:7、10:9(v/v),洗脱剂流速为30ml/min,收集洗脱液(每500ml收集一瓶),将洗脱液浓缩,根据tlc结果合瓶,依次得到7-1~7-10组份;将组份7-6上样至sephadex lh-20凝胶柱,流动相为二氯甲烷/甲醇=1:1(v/v),洗脱剂流速为0.3ml/min,收集洗脱液(每10ml收集一次),每四份洗脱液合并,依次得到7-6-1~7-6-6组份,将组份7-6-3上样至agilent c18高效液相色谱,流动性为55%甲醇水溶液,流速为3ml/min,检测器为210nm紫外光检测器,保留时间59min下,得到单体化合物1,保留时间62min下,得到单体化合物2。
[0039]
(3)结构鉴定:以氘代二甲基亚砜为溶剂,使用bruker avanceⅱ500m核磁共振仪测定分离得到的化合物1-2的核磁图谱并利用质谱推断分子式;使用圆二色谱仪j-810-150s检测化合物1-2的圆二色光谱;使用dp4+软件对化合物1-2进行空间结构计算,结合核磁图谱、cd结果与计算最终对产物结构进行表征。
[0040]
化合物1的光谱数据如下:
[0041]1h-nmr(500hz,dmso-d6)δ:3.14(1h,d,j=13.5hz,h-1a),4.17(1h,d,j=13.5hz,h-1b),9.45(1h,s,h-3oh),6.63(1h,d,j=8.0hz,h-4),7.00(1h,t,j=8.0hz,h-5),6.72(1h,d,j=8.0hz,h-6),6.56(1h,d,j=12.5hz,h-8),6.01(1h,d,j=12.5hz,h-9),3.72(1h,q,j=6.0hz,h-11),0.87(3h,d,j=6.0hz,h-12),5.04(1h,d,j=12.5hz,h-1'a),5.23
(1h,d,j=12.5hz,h-1'b),9.67(1h,s,h-3'oh),6.83(1h,d,j=8.0hz,h-4'),7.05(1h,t,j=8.0hz,h-5'),6.76(1h,d,j=8.0hz,h-6'),4.04(1h,d,j=10.0hz,h-8'),4.54(1h,d,j=10.0hz,h-9'),3.66(1h,s,h-10'),2.17(3h,s,h-12')。
[0042]
13
c-nmr(125hz,dmso-d6)δ:59.5(c-1),126.8(c-2),153.3(c-3),114.4(c-4),128.3(c-5),121.2(c-6),137.4(c-7),131.1(c-8),137.1(c-9),93.7(c-10),81.7(c-11),12.9(c-12),61.0(c-1'),122.8(c-2'),156.1(c-3'),115.3(c-4'),128.6(c-5'),123.3(c-6'),139.3(c-7'),59.8(c-8'),79.3(c-9'),79.7(c-10'),209.5(c-11'),28.0(c-12')。
[0043]
化合物1的hmbc谱结果如下:
[0044][0045]
化合物1的noesy谱结果如下:
[0046][0047]
化合物2的光谱数据如下:
[0048]1h-nmr(500hz,dmso-d6)δ:4.33(1h,d,j=13.5hz,h-1a),5.11(1h,d,j=13.5hz,h-1b),9.62(1h,s,h-3oh),6.83(1h,d,j=8.0hz,h-4),7.16(1h,t,j=8.0hz,h-5),6.85(1h,d,j=8.0hz),6.63(1h,d,j=12.5hz,h-8),5.85(1h,d,j=12.5hz,h-9),4.31(1h,q,j=6.5hz,h-11),1.31(3h,d,j=6.5hz,h-12),4.33(1h,d,j=13.5hz,h-1'a),5.27(1h,d,j=13.5hz,h-1'b),9.40(1h,s,h-3'oh),6.58(1h,d,j=7.8hz,h-4'),6.53(1h,t,j=7.8hz,h-5'),6.82(1h,d,j=7.8hz,h-6'),3.84(1h,d,j=10.0hz,h-8'),3.75(1h,d,j=10.0hz,h-9'),4.01(1h,s,h-10'),2.14(3h,s,h-12')。
[0049]
13
c-nmr(125hz,dmso-d6)δ:59.1(c-1),126.0(c-2),154.0(c-3),115.2(c-4),128.6(c-5),122.1(c-6),137.5(c-7),131.6(c-8),132.9(c-9),92.5(c-10),84.6(c-11),20.7(c-12),64.0(c-1'),126.4(c-2'),154.4(c-3'),113.8(c-4'),127.9(c-5'),119.8(c-6'),136.7(c-7'),52.5(c-8'),78.7(c-9'),91.5(c-10'),205.3(c-11'),27.7(c-12').
[0050]
化合物2的hmbc谱结果如下:
[0051][0052]
化合物2的noesy谱结果如下:
[0053][0054]
实施例2
[0055]
实施例1制备的化合物1-2的抗炎活性研究
[0056]
小鼠raw 264.7巨噬细胞的培养:将raw 264.7巨噬细胞接种在含10%胎牛血清(<0.5eu/ml)、1%青霉素和链霉素的高糖dmem培养基中,置于37℃、5%co2培养箱内培养。当细胞覆盖率达到约90%时,弃掉上清,直接加入37℃培养基吹打传代。取对数期细胞用于后续实验。
[0057]
lps诱导的raw 264.7巨噬细胞炎症模型的建立:取对数期生长的raw 264.7细胞以每孔1
×
104个细胞接种于96孔细胞培养板中,待24h细胞贴壁后,设置正常对照组、lps模型组、阳性对照组和以及化合物组,每组6个复孔。正常对照组和lps模型组先以无血清dmem培养基孵育细胞,化合物组分别以含终浓度为40.00、20.00、10.00、5μm化合物1、2的无血清dmem孵育细胞,阳性对照组以含终浓度为200μm的l-name(一氧化氮合酶抑制剂)的无血清dmem孵育细胞;按上述处理方式孵育细胞4h后,除正常对照组采用无血清dmem继续孵育外,其余各组再加入终浓度为1μg/ml lps刺激细胞,24h后收集50μl各孔培养基,运用一氧化氮检测试剂盒检测no的分泌情况,同时在每孔中加入10μl浓度为5g/l的mtt,继续培养4h后弃上清,每孔加入150μl dmso,震荡10min,在570nm处测量各孔的吸光度。
[0058]
mtt结果显示实施例1制备的化合物1-2的ic
50
》100μm,抗炎实验结果如图13所示,与模型组相比,化合物1和化合物2在各浓度下均对no的释放有不同程度的抑制,在5μm的浓度下均表现出比阳性对照组更优的抑制能力,表明化合物1与化合物2具有优良的抗炎效果。
[0059]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术
方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1