一种生产L-瓜氨酸的大肠杆菌及其构建方法和应用
一种生产l-瓜氨酸的大肠杆菌及其构建方法和应用
技术领域
1.本发明涉及一种生产l-瓜氨酸的大肠杆菌及其构建方法和应用,属于基因工程领域。
背景技术:
2.l-瓜氨酸又名氨基甲酰鸟氨酸,其分子量为175,分子式为c6h
13
o3n3,是一种非蛋白质氨基酸,溶于水,不溶于乙醇和乙醚。瓜氨酸作为天然氨基酸具有检测诊断类风湿关节炎、预防治疗心脑血管疾病、抗氧化和延缓衰老、增强肌肉力量和持久力、预防男性疾病和提高性功能等重要的生化和生理学功能,目前已在医药、工业、食品、化妆品、畜牧业等领域有广泛的应用,具有重要的经济和社会价值。
3.l-瓜氨酸的生产方法包括天然原料的提取分离、化学合成法、微生物发酵法和微生物酶法。天然原料的提取分离是指从天然原材料中通过离子交换树脂等方法将l-瓜氨酸分离提取。但由于大部分的原料瓜氨酸含量较少,导致收率很低,并且天然物中成分复杂,提取纯化工艺较为复杂,这些因素导致了提取分离法的高成本,低经济效益。化学合成法生产瓜氨酸主要有两种方法,一是在碱性条件下水解l-精氨酸;二是l-鸟氨酸盐酸盐与碱式碳酸铜反应生成二鸟氨酸合铜以保护α-nh2,用尿素甲酰化后生成l-瓜氨酸合铜,再用h2s除去铜离子。此法产率较低,环境污染大,而且生产过程中容易产生d型旋光对映体,造成后期产品分离纯化成本很高,因此化学合成法很难投入到实际应用中。微生物酶法是指使用pseudomonas putida,streptococcus faecalis等微生物生产的精氨酸脱亚胺酶和精氨酸分解酶,通过一步酶促反应分解l-精氨酸生成l-瓜氨酸,精但氨酸脱亚胺酶的酶活较低且不够稳定,不利于工业生产。微生物发酵法是通过诱变筛选出高产型突变株或者通过代谢改造模式菌株实现以廉价碳源直接发酵生产l-瓜氨酸的目的,采用发酵法生产l-瓜氨酸生产工艺相对简单、对环境影响相对较小、成本低,产量高,产物纯度高,适合大规模工业生产,在医药工业中微生物发酵法有很好的发展前景。
4.由于l-瓜氨酸合成代谢途径中存在诸多反馈调控,整个合成途径较长,并且合成l-瓜氨酸所需前体物涉及到的代谢网络复杂,因此最初利用微生物发酵法生产l-瓜氨酸的工业生产菌株主要采用传统诱变结合结构类似物抗性筛选的方法获取。研究策略集中在筛选结构类似物突变体,以解除l-瓜氨酸合成过程中的反馈调控,提高胞内l-瓜氨酸的积累量。其中,协和发酵公司(日本)使用石蜡节杆菌arthrobacter paraffineus突变菌株,发酵72h获得25.9g/l瓜氨酸。okumura shinji等经过x射线逐级诱变筛选到一株精氨酸需求型突变菌株(公开于专利us3282794),经过96h发酵培养,l-瓜氨酸氨酸的积累量为19g/l。但是经过诱变筛选得到的生产菌株,存在遗传稳定性差,容易产生回复突变等缺点很难投入到大批量工业生产中。
5.随着基因工程技术的飞速发展,运用代谢工程技术构建l-瓜氨酸生产菌株的方法逐渐替代了传统诱变育种方法。谷氨酸棒状杆菌作为传统的氨基酸发酵生产菌株,其胞内摄取的葡萄糖经过糖酵解途径生成谷氨酸的代谢流量较强,而谷氨酸作为l-瓜氨酸合成的
主要的前体物之一,因此谷氨酸棒状杆菌作为构建l-瓜氨酸生产菌株的主要选择。谢红翠等以谷氨酸棒状杆菌为出发菌株,通过除去编码阻遏蛋白的laci序列,截断乳糖阻遏蛋白来源的表达方法成功构建一株瓜氨酸生产菌株,实现合成瓜氨酸的相关基因簇argcjbdf在谷氨酸棒状杆菌中的组成型表达(谢红翠,郝宁,韦萍,等.瓜氨酸生产相关基因簇在谷氨酸棒杆菌中的组成型表达[j].南京工业大学学报:自然科学版,2011.),瓜氨酸产量为4.33g/l。现有技术中的l-瓜氨酸生产菌株存在生长周期长,生产强度低,生产易波动,菌体生长受限等问题,同时在菌株构建过程中采用将l-瓜氨酸合成关键基因连接到表达载体上以提高关键酶的转录量,以增强l-瓜氨酸合成途径的代谢流,但在生产过程中表达载体容易丢失或者需要加入一定的选择性压力,限制了菌株的工业化应用。
[0006]
大肠杆菌由于发酵周期短,遗传背景清晰,分子操作便捷和发酵工艺稳定等优势,成为构建l-精氨酸工业生产菌株的更好选择。thorben schramm等(10.1016/j.ymben.2020.03.004)以大肠杆菌为出发菌株,通过将筛选得到温度敏感型精氨酸琥珀酸合成酶(由argg基因编码)整合至基因组以解除瓜氨酸合成途径反馈调控机制,该菌株在1l发酵罐中培养45h后,瓜氨酸积累量达到3g/l。该菌株实现菌株生长与瓜氨酸生产的平衡,但在生产过程中温度变化会对菌体生理特征造成严重影响,并且该菌株瓜氨酸生产性能较弱,难以实现工业化生产。
技术实现要素:
[0007]
技术问题:大肠杆菌中l-瓜氨酸合成途径长,存在诸多反馈调控,并且合成l-瓜氨酸所需前体物涉及到的代谢网络复杂。本发明以e.coli bl21为出发菌株,使用基于crispr/cpf1的大肠杆菌多基因编辑系统对大肠杆菌进行基因的整合、无义突变,重新构建了l-瓜氨酸循环合成途径。
[0008]
技术方案:
[0009]
本发明的第一个目的是提供一种高效稳定生产l-瓜氨酸的基因工程菌株ctl,所述基因工程菌株ctl以大肠杆菌为宿主,在基因组上整合了l-瓜氨酸合成途径相关基因簇argcjbdf,抑制或降低了乙酰鸟氨酸脱乙酰基酶基因arge和精氨基琥珀酸合酶基因argg。
[0010]
在本发明的一种实施方式中,所述大肠杆菌为e.coli bl21。
[0011]
在本发明的一种实施方式中,l-瓜氨酸合成途径相关基因簇argcjbdf来源于谷氨酸棒杆菌13032。
[0012]
在本发明的一种实施方式中,所述l-瓜氨酸合成途径相关基因簇argcjbdf的核苷酸序列如seq id no.2所示。
[0013]
在本发明的一种实施方式中,所述l-瓜氨酸合成途径相关基因簇argcjbdf通过启动子控制表达。
[0014]
在本发明的一种实施方式中,所述启动子的核苷酸序列如seq id no.1所示。
[0015]
在本发明的一种实施方式中,通过无义突变抑制或降低乙酰鸟氨酸脱乙酰基酶基因arge和精氨基琥珀酸合酶基因argg。
[0016]
在本发明的一种实施方式中,通过无义突变将氨基酸序列如seq id no.9的乙酰鸟氨酸脱乙酰基酶的第12和14位氨基酸突变为终止密码子。
[0017]
在本发明的一种实施方式中,通过无义突变将氨基酸序列如seq id no.11的精氨
基琥珀酸合酶的第90、92和94位氨基酸突变为终止密码子。
[0018]
在本发明的一种实施方式中,所述l-瓜氨酸合成途径相关基因簇argcjbdf的整合位点为错配识别蛋白muts。
[0019]
本发明的第二个目的是提供一种构建所述高效稳定生产l-瓜氨酸的基因工程菌株ctl的方法,所述方法为将待编辑的基因的表达框或敲除框,及基因组同源片段连接至pcreg载体上,通过基于crispr/cpf1的基因组无痕编辑系统进行基因编辑。
[0020]
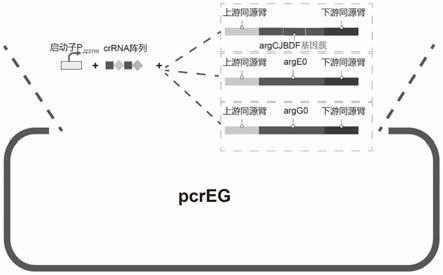
具体的该系列的质粒的构建方法、引物设计、验证引物设计均由zhu等人在combining crispr
–
cpf1 and recombineering facilitates fast and efficient genome editing in escherichia coli.acs synthetic biology,2022中的描述。
[0021]
在本发明的一种实施方式中,使用crispr/cpf1大肠杆菌基因编辑系统中描述的方法,依次将上述质粒转化至e.coli bl21中,得到最后的工程菌株e.coli ctl。
[0022]
在本发明的一种实施方式中,所述方法的具体步骤如下:
[0023]
(1)分别将待编辑的基因的表达框及基因组同源片段连接至载体pcreg,构建获得argcjbdf基因簇整合质粒pcreg-argcjbdf、pcreg-argg和pcreg-arge。
[0024]
(2)以步骤(1)中质粒pcreg-cr-argg为模板,利用引物argg0-f/r进行pcr引入终止密码子,得到回复突变质粒pcreg-argg0;以质粒pcreg-cr-arge为模板,利用引物arge0-f/r进行pcr引入终止密码子,得到回复突变质粒pcreg-arge0;
[0025]
(3)将质粒peccpf1转化到大肠杆菌e.coli bl21并制备成感受态细胞;
[0026]
(4)添加阿拉伯糖,将步骤(1)中质粒pcreg-argcjbdf、pcreg-argg和pcreg-arge依次转入步骤(3)中的感受态细胞中,得到工程菌株e.coli ctl。
[0027]
本发明还提供了一种制备l-瓜氨酸的方法,所述方法是在适宜条件下培养所述基因工程菌ctl,并从其培养物中收集l-瓜氨酸。
[0028]
在本发明的一种实施方式中,所述方法的发酵培养基组成为:葡萄糖30g/l,酵母提取物3g/l,蛋白胨3g/l,k2hpo42.5g/l,柠檬酸钠1.5g/l,mgso4·
7h2o 2.5g/l,feso4
·
7h2o 15mg/l,mnso4·
7h2o 15mg/l,vb1、vb3、vb5、vb12、vh各2mg/l,精氨酸0.1-0.5g/l,其余为水,ph7.0-7.2。
[0029]
本发明还提供了上述基因工程菌株ctl在制备含有l-瓜氨酸的产品中的应用。
[0030]
有益效果:本发明选用生长周期短、代谢途径清晰、分子操作便捷的大肠杆菌bl21为出发菌株,从l-瓜氨酸合成代谢途径的基因工程改造出发,重构了大肠杆菌中l-瓜氨酸合成途径,构建方法简单快捷,得到了一株遗传背景清晰,不携带质粒,不经诱变且能稳定高效生产l-瓜氨酸的基因工程菌株。获得的大肠杆菌e.coli ctl构建了l-瓜氨酸的外源循环路径,组成型表达使得合成途径不受内源调控系统调控,并降低l-瓜氨酸的降解,从而有效提高了l-瓜氨酸的产量。
附图说明
[0031]
图1crispr/cpf1系统质粒组构建示意图。
[0032]
图2菌株e.coli ctl摇瓶发酵数据图。
具体实施方式
[0033]
(一)菌株和载体
[0034]
质粒的构建在e.coli dh5α中进行,质粒构建完成构转化到e.coli bl21中进行整合与无义突变。所使用的载体peccpf1、peccpf1h和pcreg为zhu等人在combining crispr
–
cpf1and recombineering facilitates fast and efficient genome editing in escherichia coli.acs synthetic biology,2022中描述。
[0035]
(二)培养基
[0036]
e.coli均使用lb培养基(每升含10g胰蛋白胨、5g酵母粉和10g nacl)进行培养。
[0037]
斜面培养基组成为:葡萄糖1g/l,蛋白胨10g/l,牛肉膏10g/l,酵母粉5g/l,nacl 5g/l,琼脂25g/l,其余为水,ph 7.0-7.2;
[0038]
种子培养基组成为:葡萄糖25g/l,酵母提取物5g/l,蛋白胨3g/l,k2hpo4 1.2 g/l,mgso4
·
7h2o 0.5g/l,feso4
·
7h2o 10mg/l,mnso4
·
7h2o 10mg/l,vb1、vb3、vb5、vb12各1.3mg/l,vh 1mg/l,其余为水,ph 7.0-7.2。
[0039]
发酵培养基组成为:葡萄糖30g/l,酵母提取物3g/l,蛋白胨3g/l,k2hpo42.5 g/l,柠檬酸钠1.5g/l,mgso4·
7h2o 2.5g/l,feso4
·
7h2o 15mg/l,mnso4·
7h2o 15mg/l,vb1、vb3、vb5、vb12、vh各2mg/l,精氨酸0.1-0.5g/l,其余为水,ph 7.0-7.2。
[0040]
(三)l-瓜氨酸测定
[0041]
瓜氨酸的定量测定采用异硫氰酸苯酯将氨基酸柱前衍生后进行定量分析的高效液相色谱法,该方法参照文献(周慧等;柱前衍生化hplc法测定发酵液中l-瓜氨酸和l-鸟氨酸含量。南京工业大学学报(自然科学版).2009,31(02))。
[0042]
实施例1:crispr系列质粒的构建
[0043]
zhu等人在combining crispr
–
cpf1 and recombineering facilitates fast and efficient genome editing in escherichia coli.acs synthetic biology,2022中描述,crispr/cpf1系统由三个质粒质粒peccpf1、质粒peccpf1h和质粒pcreg组成。其中质粒peccpf1和质粒pcreg用于在大肠杆菌基因组上整合基因。质粒peccpf1为基因fncpf1和λ-red重组酶系的表达载体,含有psc101复制子并具有卡那霉素抗性。λ-red重组酶系来源于大肠杆菌的λ噬菌体,用于促进大肠杆菌中的同源重组,λ-red重组酶系通过诱导型启动子p
arab
调控表达。由于该质粒含有sacb基因,当培养基中存在蔗糖时会对细胞产生毒性,从而可以将该质粒消除。质粒pcreg为crrna的阵列表达载体,含有pmb1复制子并带有壮观霉素抗性,包括crrna阵列插入区和同源臂插入区,其中crrna阵列插入区两端含有eco31 i酶切位点,可以用于crrna的快速组装,同源臂插入区中含有sfgfp基因,可以用于同源臂插入后的快速筛选,crrna阵列插入区上游含有p
j23119
启动子,用于所需crrna阵列的表达。
[0044]
如图1所示,在进行基因操作一共需要构建2个pcreg系列的质粒,该系列质粒由质粒骨架、上游同源臂、下游同源臂、插入(替换)序列四部分组成,质粒构建方法、引物设计、验证引物设计均由zhu等人在combining crispr
–
cpf1 and recombineering facilitates fast and efficient genome editing in escherichia coli.acs synthetic biology,2022中描述。
[0045]
(一)构建质粒所用的引物
[0046]
1)argcjbdf基因簇整合质粒pcreg-argcjbdf:整合位点为大肠杆菌e.coli bl21
基因组上的muts位点,插入序列由合成启动子spl-l1(核苷酸序列为seq id no.1)和argcjbdf基因簇表达框(核苷酸序列为seq id no.2,argcjbdf基因簇的氨基酸序列分别为seq id no.3-7)组成,构建质粒所采用的引物为:
[0047]
表1 argcjbdf基因簇整合质粒pcreg-argcjbdf构建引物
[0048][0049][0050]
2)arge无义突变质粒pcreg-arge0:将e.coli基因组上arge(核苷酸序列为seq id no.8,氨基酸序列分别为seq id no.9)替换为arge0,引入数个终止密码子以失活arge。
[0051]
表2 arge无义突变质粒pcreg-arge0构建引物
[0052][0053]
3)argg回复突变质粒pcreg-argg0:将e.coli基因组上argg(核苷酸序列和氨基酸序列分别如seq id no.10和seq id no.11)替换为,引入数个终止密码子以失活argg。
[0054]
表3 argg无义突变质粒pcreg-argg0构建引物
[0055][0056]
4)其他验证引物如下:
[0057]
表4质粒验证引物
[0058][0059][0060]
(二)质粒构建方法
[0061]
1)crrna连接
[0062]
单个crrna的连接:此时crrna可以直接通过设计带有重叠区的一对引物,经过变性退火形成带有粘性末端的引物二聚体。使用碧云天的5
×
annealing buffer for dna oligos,引物浓度为10μm,50μl体系中上下游引物(cr-基因-f/r,基因分别为muts、arge、argg)各20μl,annealing buffer 10μl。反应条件为:98℃2min,0.1℃/s降温至4℃后保温。将二聚体稀释10倍,取1μl与eco31 i酶切后的载体pcreg连接,转入e.coli dh5α感受态中,使用验证引物sfgfp-r和cr-基因-f(基因为muts、arge、argg)进行菌落pcr验证,获得3个质粒pcreg-cr-argcjbdf、pcreg-cr-argg、pcreg-cr-arge。
[0063]
2)同源臂的扩增与连接
[0064]
使用cjbdf-500uf和cjbdf-500ur,以大肠杆菌e.coli bl21基因组作为模板,扩增整合位点muts的上游同源臂;使用cjbdf-500df和cjbdf-500dr,以大肠杆菌e.coli bl21基因组作为模板,扩增整合位点muts的下游同源臂。使用cjbdf-f/r从合成的基因(核苷酸序列如seq id no.12)上扩增插入序列;使用argg-f/r,arge-f/r以大肠杆菌e.coli bl21基因组作为模板,分别扩增基因argg和arge的同源臂,pcr产物用dna纯化试剂盒回收。
[0065]
使用引物pcreg-lif和pcreg-lir对步骤1)中的3个质粒pcreg-cr-argcjbdf、pcreg-cr-argg和pcreg-cr-arge进行线性化,得到线性化载体。
[0066]
使用碧云天seamless cloning kit(无缝克隆试剂盒)分别将3个质粒的线性化载体、上下游同源臂、插入(替换)基因片段(argcjbdf基因簇基因片段)连接后,分别转入e.coli dh5α感受态中,获得转化子。使用j23119-f和hominsert-r对转化子进行验证,测序,获得质粒pcreg-argcjbdf、pcreg-arge和pcreg-argg。
[0067]
使用arge0-f/r和argg0-f/r分别对测序正确质粒pcreg-arge和pcreg-argg进行pcr,引入终止密码子,将pcr产物转入e.coli dh5α感受态中。使用j23119-f和argg-er/arge-gr对转化子进行验证,测序,得到回复突变质粒pcreg-arge0和pcreg-argg0。
[0068]
实施例2:基因工程菌株ctl的构建
[0069]
使用实施例1中构建的质粒对大肠杆菌进行基因编辑,包括如下步骤(后面所述的抗生素浓度为:卡那霉素:50μg/ml,壮观霉素:100μg/ml):
[0070]
(1)首先将cpf1表达载体peccpf1转化到大肠杆菌e.coli bl21中,涂布在含有卡那霉素的lb培养基平板上得到阳性菌落。
[0071]
(2)接菌培养,制备含有质粒peccpf1的大肠杆菌感受态,加入终浓度为10mm的阿拉伯糖诱导质粒peccpf1上λ-red同源重组酶的表达,将整合质粒pcreg-argcjbdf加入含有质粒peccpf1的大肠杆菌感受态中,冰浴30min后42℃热激90s,加入1ml的lb培养基后培养1.5-2h,然后5000g离心3分钟后弃去900μl培养基,留下100μl培养基将菌体重悬并涂布到含有卡那霉素和壮观霉素的lb平板上。
[0072]
(3)长出单菌落后通过菌落pcr(引物muts-gf和cjbdf-r)验证基因编辑情况,阳性菌株可以只将第二个质粒(即crrna表达质粒pcreg-argcjbdf)消除,而将peccpf1质粒留在菌中,重复上述操作,将pcreg-arge0、pcreg-argg0依次转入大肠杆菌,使用argg-gf与argg-gr验证arge是否插入终止密码子,使用arge-gf和arge-gr验证argg是否插入终止密码子。
[0073]
(4)最终得到消除质粒的基因工程菌株e.coli ctl。质粒消除方法由a modified pcas/ptargetf system for crispr-cas9-assisted genome editing in escherichia coli(doi:10.1093/abbs/gmab036)中描述。
[0074]
实施例3:基因工程菌株e.coli ctl l-瓜氨酸的生产验证
[0075]
基因工程菌株e.coli ctl与e.coli bl21摇瓶发酵结果对比如图2所示,以验证重构l-瓜氨酸合成途径对l-瓜氨酸积累的效果。
[0076]
斜面培养:取-80℃保藏菌种划线接种于活化斜面,37℃培养12h,并传代一次;
[0077]
摇瓶种子培养:用接种环刮取一环斜面种子接种于装有30ml种子培养基的500ml三角瓶中,九层纱布封口,37℃,200rpm培养8h;
[0078]
摇瓶发酵培养:按种子培养液体积10%的接种量接种到装有发酵培养基的500ml三角瓶中(终体积为30ml),九层纱布封口,37℃,200r/min振荡培养,发酵过程中通过补加氨水维持ph在7.0-7.2;补加60%(m/v)葡萄糖溶液维持发酵进行;发酵周期28h;
[0079]
经过36h摇瓶发酵,e.coli bl21菌株od600为52,发酵液中无l-瓜氨酸积累,重组菌株e.coli ctl od600为45,发酵液中l-瓜氨酸积累量达到5.6g/l。与对照菌株相比,重组菌株e.coli ctl od600略有点下降,但外源引入的l-瓜氨酸循环合成途径能够有效的合成l-瓜氨酸。
[0080]
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1