一种鉴别菊叶薯蓣超雄株的引物对、试剂盒及其筛选方法和应用与流程
1.本发明属于菊叶薯蓣育种技术领域,尤其涉及一种鉴别菊叶薯蓣超雄株的引物对、试剂盒及其筛选方法和应用。
背景技术:
2.菊叶薯蓣(dioscorea composita hemsl.)是薯蓣科薯蓣属植物。多年生缠绕性草本。地下茎有的形如掌状,有的棒状迭生;外皮粗糙,黑褐色,分背腹两面;根系完全布于腹。块茎上有芽眼,肉眼难以识别。地上茎圆形肉质,直径0.4-0.7厘米。叶为单叶互生;叶连柄长25-30厘米、宽10-15厘米;主脉突起11-13条;叶面皱摺或平滑,心脏楔形或卵圆形或椭圆形渐尖成尾状;叶柄紫色,或浅或深;叶终年常绿。
3.菊叶薯蓣为雌、雄异株,雄株因不结种子而消耗养分少,产量和皂苷含量比同条件下相同的雌株高。但有关薯蓣雌雄性别决定机制的研究相当困乏,且在自然群体中常常出现雄性植株产生两性花,引起自花或异花授粉。因此寻找菊叶薯蓣超雄株,对于开展菊叶薯蓣的全雄育种十分必要;但是目前并没有快速高效的鉴别菊叶薯蓣超雄株的方法。
技术实现要素:
4.有鉴于此,本发明的目的在于提供一种能够鉴别菊叶薯蓣超雄株的引物对、试剂盒及其应用。
5.本发明提供了一种鉴别菊叶薯蓣超雄株的引物对,包括上游引物和下游引物;所述上游引物的核苷酸序列如seq id no.1所示,所述下游引物的序列如seq id no.2所示。
6.本发明提供了一种鉴别菊叶薯蓣超雄株的试剂盒,包括所述的引物对和检测试剂。
7.优选的,所述检测试剂包括pcr扩增试剂。
8.本发明还提供了所述的引物对、所述的试剂盒在菊叶薯蓣全雄育种中的应用。
9.优选的,所述应用为利用所述的引物对对待检测的菊叶薯蓣的基因组dna进行pcr扩增,若扩增产物有且仅有159bp的片段,则待检测菊叶薯蓣为超雄株,否则不是超雄株。
10.本发明还提供了鉴别菊叶薯蓣超雄株的引物对的筛选方法,包括以下步骤:
11.1)将菊叶薯蓣雌、雄株各3个生物学重复进行转录组学测序获得雌株转录本测序数据和雄株转录本测序数据;
12.2)比较步骤1)中所述的雌株转录本数据和雄株转录本数据,筛选在雄株转录本数据中高表达、并且在雌株转录本数据中低表达或不表达的ssr位点;
13.3)根据步骤2)筛选获得的ssr位点设计初始扩增引物,
14.4)将设计获得的初始扩增引物进行人工修改获得兼并引物;所述人工修改为保留初始扩增引物中上游引物的第5至第19位碱基,将初始扩增引物中的下游引物的第9、11、13、15的碱基替换为n;
15.5)分别以菊叶薯蓣雌、雄株的基因组dna为模板,以步骤4)获得的兼并引物进行热不对称pcr扩增获得扩增产物;
16.6)将所述扩增产物进行电泳,挑选菊叶薯蓣雌、雄株扩增产物的差异条带;
17.7)将步骤6)中获得的差异条带回收,并测序获得菊叶薯蓣雌、雄株差异序列,根据所述菊叶薯蓣雌、雄株差异序列设计获得二轮扩增引物;
18.8)分别以菊叶薯蓣雌、雄株的基因组dna为模板,以步骤7)获得的二轮扩增引物,进行第二轮pcr扩增,获得二轮扩增产物;
19.9)将所述二轮扩增产物进行电泳,挑选菊叶薯蓣雌、雄株扩增产物的差异条带;
20.10)将步骤9)中获得的差异条带回收,并测序获得菊叶薯蓣雌、雄株差异序列,根据所述菊叶薯蓣雌、雄株差异序列设计获得三轮扩增引物,即为鉴别菊叶薯蓣超雄株的引物对。
21.优选的,步骤1)中的菊叶薯蓣雌、雄株为6年生以上的菊叶薯蓣雌、雄株。
22.与现有技术相比,本发明具有如下有益效果:本发明提供的鉴别菊叶薯蓣超雄株的引物对是通过筛选在雄株转录本数据中高表达、并且在雌株转录本数据中低表达或不表达的ssr位点并设计兼并引物经过多轮扩增筛选后获得的,能够快速高效的鉴别菊叶薯蓣雌雄株,并且对于不同质量的模板dna均能够实现稳定准确的鉴定。
附图说明
23.图1为由初始引物经人工修改获得的25对兼并引物的序列;
24.图2为8株雌性、4株雄性共12个模板的热不对称pcr扩增产物的电泳结果;其中f1~f6为雌株模板,m1~m4为雌株模板;marker的分子量大小为5000bp;
25.图3为第二轮pcr扩增产物电泳结果,其中m1~m3为雄株模板,f1~f5为雌株模板,marker的分子量大小为5000bp;
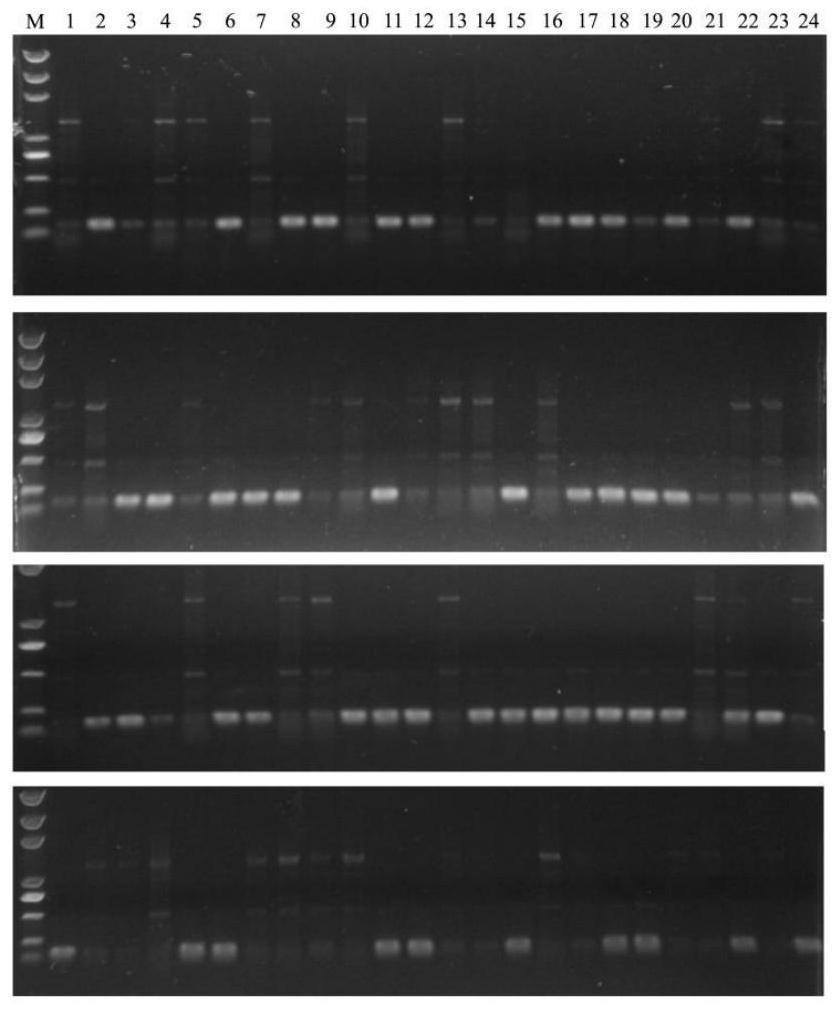
26.图4为第三轮pcr扩增产物电泳结果,其中1~12为雌株模板,13~24为雄株模板;marker的分子量大小为5000bp;
27.图5为以所筛选到的引物对扩增不同质量的dna模板获得的扩增产物的电泳结果,其中1~16为不同质量的dna模板;marker的分子量大小为5000bp;
28.图6为以所筛选到的引物对鉴定96株未知性别的菊叶薯蓣材料的结果;
29.图7为对比例rapd的引物扩增结果;其中f1~f6为雌株模板,m1~m4为雌株模板;marker的分子量大小为5000bp;
30.图8为对比例sarp的扩增结果;其中f1~f6为雌株模板,m1~m4为雌株模板;marker的分子量大小为5000bp;
31.图9为对比例issr的扩增结果:其中f1~f6为雌株模板,m1~m4为雌株模板;marker的分子量大小为5000bp;
32.图10为ssr分子标记引物扩增结果;
33.图11为第一次差异条带挖带回收后没有差异的引物扩增结果;
34.图12为为第二次挖带回收后没有差异的引物扩增结果。
具体实施方式
35.本发明提供了一种鉴别菊叶薯蓣超雄株的引物对,包括上游引物和下游引物;所述上游引物的核苷酸序列如seq id no.1所示,所述下游引物的序列如seq id no.2所示;具体如下:
36.上游引物(5
’→3’
):gggtttcttcccttcgctgc(seq id no.1)
37.下游引物(5
’→3’
):gagatggaaacccaaatgctgac(seq id no.2)。
38.引物性能参数见表1。
39.表1引物性能参数
[0040] ratingseqnolengthtm(℃)gc%δg[kcals/mol]activitydegeneracytaoptsense1003982063.560.0-42,836.91
‑‑
anti-sense1005562362.547.8-43.130.71
‑‑
product84
‑‑
15984.945.9
‑‑‑‑‑‑
53.2
[0041]
本发明提供了一种鉴别菊叶薯蓣超雄株的试剂盒,包括所述的引物对和检测试剂。
[0042]
在本发明中,所述检测试剂优选的包括pcr扩增试剂。本发明对所述pcr扩增试剂的种类和规格没有特殊限定,采用本领域常规市售的pcr扩增试剂能够实现扩增即可。
[0043]
本发明还提供了所述的引物对、所述的试剂盒在菊叶薯蓣全雄育种中的应用。
[0044]
在本发明中,所述应用为利用所述的引物对对待检测的菊叶薯蓣的基因组dna进行pcr扩增,若扩增产物有且仅有一条159bp的片段,则待检测菊叶薯蓣为超雄株,否则不是超雄株。在本发明具体实施过程中,所述pcr扩增的体系以25μl计,包括如下组成:2
×
taq mix预混液12.5μl,上游引物1μl,下游引物1μl,模板1μl,ddh2o 9.5μl。
[0045]
所述pcr扩增的程序如下:预变性94℃,2min;变性94℃,15s,退火55℃,15s,延伸72℃,15s;变性退火延伸30个循环,72℃保持2min。
[0046]
本发明还提供了鉴别菊叶薯蓣超雄株的引物对的筛选方法,包括以下步骤:
[0047]
1)将菊叶薯蓣雌、雄株材料进行转录组学测序获得雌株转录本测序数据和雄株转录本测序数据;
[0048]
2)比较步骤1)中所述的雌株转录本数据和雄株转录本数据,筛选在雄株转录本数据中高表达、并且在雌株转录本数据中低表达或不表达的ssr位点;
[0049]
3)根据步骤2)筛选获得的ssr位点设计初始扩增引物,
[0050]
4)将设计获得的初始扩增引物进行人工修改获得兼并引物;所述人工修改为保留初始扩增引物中上游引物的第5至第19位碱基,将初始扩增引物中的下游引物的第9、11、13、15的碱基替换为n;
[0051]
5)分别以菊叶薯蓣雌、雄株的基因组dna为模板,以步骤4)获得的兼并引物进行热不对称pcr扩增获得扩增产物;
[0052]
6)将所述扩增产物进行电泳,挑选菊叶薯蓣雌、雄株扩增产物的差异条带;
[0053]
7)将步骤6)中获得的差异条带回收,并测序获得菊叶薯蓣雌、雄株差异序列,根据所述菊叶薯蓣雌、雄株差异序列设计获得二轮扩增引物;
[0054]
8)分别以菊叶薯蓣雌、雄株的基因组dna为模板,以步骤7)获得的二轮扩增引物,进行第二轮pcr扩增,获得二轮扩增产物;
[0055]
9)将所述二轮扩增产物进行电泳,挑选菊叶薯蓣雌、雄株扩增产物的差异条带;
[0056]
10)将步骤9)中获得的差异条带回收,并测序获得菊叶薯蓣雌、雄株差异序列,根据所述菊叶薯蓣雌、雄株差异序列设计获得三轮扩增引物,即为鉴别菊叶薯蓣超雄株的引物对。
[0057]
在本发明中,所述菊叶薯蓣雌、雄株优选为6年生以上的菊叶薯蓣雌、雄株。在本发明中,所述6年生以上的菊叶薯蓣雄株为连续6年以上表现为雄株的超雄株,不曾出现过两性花。
[0058]
本发明对所述转录组学测序和数比对分析等步骤没有特殊限定,采用本领域常规方法即可。在本发明中,在雄株转录本数据中高表达、并且在雌株转录本数据中低表达或不表达的筛选标准为首先选择差异表达log2foldchange》2的ssr位点数据,然后再从这些数据中选择雄株中组装的base mean》2000,雌株中base mean《20的ssr位点数据。
[0059]
在本发明中,人工修改获得兼并引物如图1所示。
[0060]
在本发明中,所述热不对称pcr的体系以25μl计,包括:2
×
taq mix预混液12.5μl,上游引物1μl,下游引物1μl,模板1μl,ddh2o 9.5μl。
[0061]
所述热不对称pcr的程序如下:94℃,2min;94℃15s,36℃15s,10个循环;72℃30s,94℃15s,60℃15s,20个循环;72℃30s,75℃5min。
[0062]
在本发明中,差异条带回收,测序优选的采用ta克隆测序。
[0063]
在本发明中,第二轮pcr扩增的体系以25μl计,包括:2
×
taq mix预混液12.5μl,上游引物1μl,下游引物1μl,模板1μl,ddh2o 9.5μl。
[0064]
所述第二轮pcr扩增的程序为:预变性94℃,2min;变性94℃,15s,退火55℃,15s,延伸72℃,30s;变性退火延伸30个循环;72℃保持2min。
[0065]
在本发明中,第三轮pcr扩增的体系以25μl计,包括:2
×
taq mix预混液12.5μl,上游引物1μl,下游引物1μl,模板1μl,ddh2o 9.5μl。
[0066]
所述第三轮pcr扩增的程序为:预变性94℃,2min;变性94℃,15s,退火55℃,15s,延伸72℃,15s;变性退火延伸30个循环;72℃保持2min。
[0067]
下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
[0068]
实施例1
[0069]
标记的获取过程:
[0070]
(1)将6年生菊叶薯蓣雌雄株各3个生物学重复进行转录组学测序,样本为根状茎。通过denovo拼接序列,根据ssr位点设计相关引物(由转录组测序公司提供)。按照转录本在雌雄株中差异不同筛选ssr引物(挑选转录本在雄株高表达并在雌株中低表达或不表达,具体为首先选择差异表达log2foldchange》2的ssr位点数据,然后再从这些数据中选择雄株中组装的base mean》2000,并且雌株中base mean《20的ssr位点数据。),将引物加以修改并合成,修改原则为保留上游引物的第5至第19位碱基,下游引物的第9、11、13、15的碱基替换为n,设计为兼并碱基序列,例如trinity_dn18_c1_g1:
[0071]
ttgccttgttgggtg(seq id no.99)
[0072]
cagtcgagnanancntaggg(seq id no.100)
[0073]
设计的兼并引物的序列详见图1所示。
[0074]
将上述兼并引物进行热不对称pcr,模板选择8株雌性、4株雄性共12个模板。
[0075]
热不对称pcr的体系为:2
×
taq mix预混液12.5μl,上游引物1μl,下游引物1μl,模板1μl,ddh2o 9.5μl。
[0076]
热不对称pcr的程序如下:94℃,2min;94℃15s,36℃15s,10个循环;72℃30s,94℃15s,60℃15s,20个循环;72℃30s,75℃5min。
[0077]
将热不对称pcr的扩增产物经过1%的琼脂糖凝胶电泳,结果如图2所示,挑选雄株模板和雌株模板的差异条带。
[0078]
(2)将差异条带挖带回收,经ta克隆测序,并根据测序获得的序列设计引物。再次以3株雄性、5株雌性为模板进行第二轮pcr。
[0079]
差异条带的序列如seq id no.3所示,根据改差异条带设计引入的序列如seq idno.4和seq idno.5所示。
[0080]
第二轮pcr扩增的体系为:2
×
taq mix预混液12.5μl,上游引物1μl,下游引物1μl,模板1μl,ddh2o 9.5μl。
[0081]
所述第二轮pcr扩增的程序为:预变性94℃,2min;变性94℃,15s,退火55℃,15s,延伸72℃,30s;变性退火延伸30个循环;72℃保持2min。
[0082]
将产物经1%琼脂糖电泳,结果如图3所示。
[0083]
(3)将二轮的扩增产物的差异条带再次挖带回收,并测序。根据测序获得的序列设计引物。进行第三轮扩增。
[0084]
差异条带序列如seq id no.6所示。
[0085]
根据改差异条带设计引入的序列如seq id no.7和seq id no.8所示。
[0086]
在本发明中,第三轮pcr扩增的体系为:2
×
taq mix预混液12.5μl,上游引物1μl,下游引物1μl,模板1μl,ddh2o 9.5μl
[0087]
所述第三轮pcr扩增的程序为:预变性94℃,2min;变性94℃,15s,退火55℃,15s,延伸72℃,15s;变性退火延伸30个循环;72℃保持2min。
[0088]
第三轮扩增产物电泳结果如图4所示,最终筛选获得一对引物在雄株中仅能够稳定的扩增出一条159bp的产物,
[0089]
产物序列如seq idno.9所示。
[0090]
根据该产物设计的引物序列如下:
[0091]
上游引物(5
’→3’
):gggtttcttcccttcgctgc(seq id no.1)
[0092]
下游引物(5
’→3’
):gagatggaaacccaaatgctgac(seq idno.2)。
[0093]
(4)引物扩增的效率分析。基本信息如表1所示。利用定量pcr检测到扩增效率为98.7%,r2为99.88%。
[0094]
(5)引物验证。为保证该引物具有较强的扩增效率,以不同质量的dna为模板进行验证,dna样本的od260/230对于dna本身质量影响较大,从而会直接导致pcr扩增的失败。结果如图5和表2所示,显示od260/230基本不影响实验结果。
[0095]
表2 dna样本质量情况表
[0096][0097]
(6)应用:随机挑选96株未知性别的菊叶薯蓣材料,提取基因组dna后,利用上述引物进行鉴定。由扩增结果如图6所示,可知该引物可以鉴定99%以上的菊叶薯蓣雌雄株。
[0098]
对比例
[0099]
以不同序列的引物分别对菊叶薯蓣雌雄株的基因组dna进行扩增,结果如图7~12所示,可见本发明提供的引物对相对于其他引物对能够很好的鉴别菊叶薯蓣雌雄株。
[0100]
图7为rapd分子标记引物扩增结果;
[0101]
体系:2
×
taq mix预混液12.5μl,上游引物1μl,下游引物1μl,模板1μl,ddh2o 9.5μl
[0102]
程序:94℃2min;94℃45s,36℃60s,40个循环;72℃90min;72℃5min。
[0103]
引物s1465、s2005、s2125、s1305、s1145、s1505、s2065、s2160、s1425、s1385如seq id no.10~19所示。
[0104]
图8为srap分子标记引物扩增结果
[0105]
体系:2
×
taq mix预混液12.5μl,上游引物1μl,下游引物1μl,模板1μl,ddh2o 9.5μl;
[0106]
扩增程序:94℃5min;94℃1min,36℃30s,10个循环;72℃1min;94℃1min,50℃30s,72℃90s,30个循环;72℃10min。引物em-1、me-1、me-2、me-3、me-4、me-5、me-6、me-7、me-8、me-9、me-10的序列如seq id no.20~30所示。
[0107]
图9为issr分子标记引物扩增结果
[0108]
体系:2
×
taqmix预混液12.5μl,上游引物1μl,下游引物1μl,模板1μl,ddh2o 9.5μl
[0109]
程序:94℃2min;94℃1min,55℃40s,72℃90s,40个循环;72℃10min。
[0110]
引物issr4、issr8、issr12、issr15、issr16、issr20如seq id no.31~36所示。
[0111]
图10为实施例1中设计的众多兼并引物没有扩增出差异条带的扩增结果。
[0112]
体系:2
×
taqmix预混液12.5μl,上游引物1μl,下游引物1μl,模板1μl,ddh2o 9.5μl
[0113]
程序:94℃2min;94℃15s,55℃15s,72℃15s,30个循环;72℃2min。
[0114]
引物序列:
[0115]
trinity_dn8580_c0_g1、trinity_dn45675_c0_g1、trinity_dn156139_c0_g1、trinity_dn111475_c0_g1、trinity_dn254_c0_g1、trinity_dn81195_c0_g1的序列如seq id no.37~48所示。
[0116]
图11为实施例1中第一次差异条带挖带回收后没有差异的引物扩增结果。
[0117]
体系:2
×
taq mix预混液12.5μl,上游引物1μl,下游引物1μl,模板1μl,ddh2o 9.5μl
[0118]
程序:94℃2min;94℃15s,55℃15s,72℃15s,30个循环。
[0119]
引物0-5-f、0-5-r、1-23-f、1-23-r、2-90-f、2-90-r、3-22-f、3-22-r的序列如seq id no.49~56所示。
[0120]
图12为第二次差异条带挖带回收设计的未有差异的引物扩增结果,共计21对引物;每一对引物雌雄模板总数为6,前三个泳道模板为雄株,后三个泳道模板为雌株,即除去marker泳道,每6个相邻泳道为一组,用相同的引物扩增;按照从上至下,从左至右的顺序数第15组是空白。
[0121]
体系:2
×
taq mix预混液12.5μl,上游引物1μl,下游引物1μl,模板1μl,ddh2o 9.5μl
[0122]
程序:94℃2min;94℃15s,55℃15s,72℃15s,30个循环。
[0123]
21对引物的序列如seq id no.57~98所示。
[0124]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1