一种雷替曲塞杂质及其制备方法、应用与流程
1.本发明涉及医药领域,尤其涉及一种雷替曲塞杂质及其制备方法、应用。
背景技术:
2.雷替曲塞是一种胸腺核苷酸合成酶抑制剂,用于治疗晚期直肠结肠癌患者。其化学名为:(5-(甲基((2-甲基-4-氧代-3,4-二氢喹唑啉-6-基)甲基)氨基)噻吩-2-羰基)-l-谷氨酸,结构式如下式ix所示:雷替曲塞可以由式ii所示的化合物与l-谷氨酸二乙酯盐酸盐(下式iv所示)为原料,在碱性条件下,通过酰胺缩合得到式a所示化合物,化合物a再经脱boc反应得到式b所示化合物。化合物b和下式vii所示化合物反应得到式c所示化合物,化合物c碱性条件下酯水解得到雷替曲塞。
3.雷替曲塞的合成路线如下:雷替曲塞杂质对于雷替曲塞的质量研究极为重要,目前已发现有多种雷替曲塞杂质,例如:专利cn201710601520.1中公开了七种雷替曲塞杂质a-f,结构式如下:
专利cn201710810458.7中公开了一种雷替曲塞杂质g,结构式如下:专利cn201710588226.1中公开了一种雷替曲塞杂质,结构式如下:然而,除了包括上述化合物在内的已知雷替曲塞杂质之外,仍存在一些难以被检测发现或分离的未知雷替曲塞杂质,为了进一步提高雷替曲塞的品质,有必要尽量减少雷替曲塞中未知杂质的含量。申请人在生产过程中发现了一种新的雷替曲塞杂质,该杂质未在现有任何文献中报道,并且我们进一步发现该杂质在制备雷替曲塞的过程中含量高且很难除去,亟需杂质对照品对雷替曲塞进行质量研究。因此,需要研发出一种可简单、高效合成出该杂质的方法。
技术实现要素:
4.本发明所要解决的第一个技术问题是针对现有技术中雷替曲塞存在未知杂质的现状,提供一种新的雷替曲塞杂质,该杂质可为雷替曲塞的质量研究工作提供参考和条件。
5.本发明所要解决的第二个技术问题是针对现有技术中尚无上述雷替曲塞杂质合成方法的现状,提供一种上述雷替曲塞杂质的合成方法,该合成方法简单,收率及纯度高。
6.本发明的具体技术方案为:第一方面,本发明提供了一种雷替曲塞杂质,结构式如式i所示:
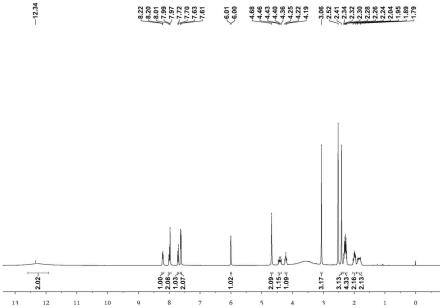
上述雷替曲塞杂质的1h-nmr图谱参数为:1h nmr(400mhz,dmso-d6):12.34(br,2h),8.21(d,j=8.0hz,1h),8.01-7.97(m,2h),7.71(d,j=8.0hz,1h),7.63-7.61(m,2h),6.01(d,j=4.3hz,1h),4.68(s,2h),4.46-4.36(m,1h),4.25-4.19(m,1h),3.17(s,3h),2.41(s,3h),2.34-2.24(m,4h),2.04-1.95(m,2h),1.89-1.79(m,2h)。
7.13
c-nmr图谱参数为:
13
c nmr(101mhz,dmso-d6):δ174.2,173.9,162.0,161.8,158.5,155.0,154.6,138.8,135.8,135.4,135.0,130.8,129.7,129.6,126.8,125.7,125.2,120.9,120.8,61.7,52.2,44.8,33.3,30.9,26.4,21.7。esi-ms m/z=588.2(m+h
+
),610.2(m+na
+
),586.3(m-h-)。
8.第二方面,本发明提供了一种如上述雷替曲塞杂质的合成方法,包括以下步骤:(1)如式ii所示的化合物依次与如式iii、式iv所示的化合物进行反应,生成如式v所示的化合物:(2)如式v所示的化合物脱除boc保护基得到如式vi所示的化合物:
(3)如式vi所示的化合物与如式vii所示的化合物反应,得到如式viii所示的化合物:(4)如式viii所示的化合物酯基水解得到如式i所示的雷替曲塞杂质:
9.作为优选,步骤(1)中:反应条件为:以有机溶剂为反应介质,以2-羟基吡啶-n-氧化物/1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐为缩合剂,以碱性化合物为敷酸剂,反应温度为30~50℃。
10.作为优选,步骤(1)中:将反应后所得反应液浓缩,加入水、有机萃取剂萃取,分去水层得到有机层,有机层水洗后减压浓缩得到如式v所示的化合物。
11.作为优选,步骤(2)中:反应条件为:以有机溶剂为反应介质,在酸-有机溶剂溶液的存在下。
12.作为优选,步骤(2)中:反应完毕后使用碱调节ph至弱碱性(优选ph=7-8),加入有机萃取剂/水萃取,分去水层得到有机层,有机层水洗后减压浓缩得到如式vi所示的化合物。
13.作为优选,步骤(3)中:反应条件为:以有机溶剂为反应介质,以酸性化合物为敷酸剂,反应温度为40~65℃。
14.作为优选,步骤(3)中:反应完毕后浓缩除去有机溶剂,加入水、有机萃取剂萃取,分去水层得到有机层,有机层水洗并分离纯化得到如式viii所示的化合物。
15.作为优选,步骤(4)中:反应条件为:以水为反应介质,在碱(优选氢氧化钠)存在下。
16.作为优选,步骤(4)中:反应完毕后,加酸调节ph析出固体,抽滤后,滤饼干燥得到雷替曲塞杂质。
17.第三方面,本发明提供了上述雷替曲塞杂质在雷替曲塞质量检测中作为杂质对照品的应用。
18.与现有技术对比,本发明的有益效果是:
(1)本发明团队在生产过程中偶然发现了一种现有文献中未报道过的新型雷替曲塞杂质,该杂质在雷替曲塞合成中含量较高,且精制效果较差。为此本发明研究了该杂质的产生途径,并定向合成该杂质,该合成工艺具有原料易得、合成步骤较短、操作简单、收率和纯度高的优点。
19.(2)本发明合成所得的新型雷替曲塞杂质对于雷替曲塞的质量研究工作有重要意义;其可作为对照品对雷替曲塞进行质量控制,应用前景较好。
附图说明
20.图1为本发明实施例1中式i所示雷替曲塞杂质的1h nmr图谱(400mhz,dmso-d6);图2为本发明实施例1中式i所示雷替曲塞杂质的
13
c nmr图谱(101mhz,dmso-d6);图3为本发明实施例1中式i所示雷替曲塞杂质的esi-ms图谱。
具体实施方式
21.下面结合实施例对本发明作进一步的描述。
22.总实施例一种雷替曲塞杂质,结构式如式i所示:一种如上述雷替曲塞杂质的合成方法,包括以下步骤:(1)如式ii所示的化合物依次与如式iii、式iv所示的化合物进行反应,生成如式v所示的化合物:步骤(1)中:反应条件为:以乙腈为反应介质,以2-羟基吡啶-n-氧化物/1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐为缩合剂,以三乙胺为敷酸剂,反应温度为30~50℃。将
反应后所得反应液浓缩,加入水、乙酸乙酯萃取,分去水层得到乙酸乙酯层,乙酸乙酯层水洗后减压浓缩得到如式v所示的化合物。
23.(2)如式v所示的化合物脱除boc保护基得到如式vi所示的化合物:步骤(2)中:反应条件为:以乙酸乙酯为反应介质,在氯化氢-乙酸乙酯溶液的存在下。反应完毕后使用碱调节ph至弱碱性(优选ph=7-8),加入乙酸乙酯/水萃取,分去水层得到乙酸乙酯层,乙酸乙酯层水洗后减压浓缩得到如式vi所示的化合物。
24.(3)如式vi所示的化合物与如式vii所示的化合物反应,得到如式viii所示的化合物:步骤(3)中:反应条件为:以乙腈为反应介质,以2,6-二甲基吡啶为敷酸剂,反应温度为40~65℃。反应完毕后浓缩除去乙腈,加入水、二氯甲烷萃取,分去水层得到二氯甲烷层,二氯甲烷层水洗并分离纯化得到如式viii所示的化合物。
25.(4)如式viii所示的化合物酯基水解得到如式i所示的雷替曲塞杂质:
26.步骤(4)中:反应条件为:以水为反应介质,在碱(优选氢氧化钠)存在下。反应完毕后,加酸调节ph析出固体,抽滤后,滤饼干燥得到雷替曲塞杂质。
27.上述雷替曲塞杂质在雷替曲塞质量检测中可作为杂质对照品应用。
28.实施例1
本实施例中雷替曲塞杂质的合成路线如下:本实施例中雷替曲塞杂质的合成路线如下:本实施例中雷替曲塞杂质的合成方法具体如下:(1)向250ml反应瓶中依次加入100ml乙腈、5g化合物ii、2.2g的hopo和3.7g的edci,35℃搅拌反应30分钟,而后加入3.4g化合物iii、2.4g三乙胺,35℃搅拌反应4小时。然后向所得反应液中再次加入2.2g的hopo和3.7g的edci,35℃搅拌反应30分钟,继续加入2.8g化合物iv、2.4g三乙胺,35℃搅拌反应4小时。减压浓缩反应液,加入乙酸乙酯/水萃取,有机相水洗后减压浓缩干得到8.7g黄色油状物v,收率75%。
29.(2)向100ml反应瓶中加入8.7g化合物v,加入25ml乙酸乙酯溶解,然后滴加16.3ml氯化氢-乙酸乙酯溶液(4m),30℃搅拌反应过夜。将反应液冷至20℃以下,滴加饱和碳酸钠溶液调节ph至8,加入乙酸乙酯/水萃取,有机相水洗后减压浓缩干,得5.8g黄色油状物vi,收率80%。
30.(3)向250ml反应瓶中依次加入100ml乙腈、5.8g化合物vi、3.2g化合物vii和1.4g的2,6-二甲基吡啶,50℃保温反应48小时。减压浓缩反应液,加水/二氯甲烷萃取,水洗有机相,浓缩有机相,反应柱层析纯化,二氯甲烷∶甲醇体积比40∶1为洗脱液,收集目标组分后浓缩干,得到4.9g类白色固体viii,收率63%。
31.(4)向100ml反应瓶中依次加入25ml水、1.5g氢氧化钠,搅拌溶清后加入4.9g化合物viii,25℃反应2小时,缓慢滴加稀盐酸,调节ph至2~3,析出固体,抽滤,滤饼真空烘干,得到3.8g淡黄色固体雷替曲塞杂质i,收率90%。如图1~3为化合物i的表征数据。
32.实施例2(1)向250ml反应瓶中依次加入100ml四氢呋喃、4.5g化合物ii、2.0g的hopo和3.3g的edci,45℃搅拌反应25分钟,而后加入3.1g化合物iii、2.2g三乙胺,40℃搅拌反应3小时。然后向所得反应液中再次加入2.0g的hopo和3.3g的edci,40℃搅拌反应25分钟,继续加入2.5g化合物iv、2.2g三乙胺,40℃搅拌反应3小时。减压浓缩反应液,加入二氯甲烷/水萃取,有机相水洗后减压浓缩干得到7.1g黄色油状物v,收率68%。
33.(2)向100ml反应瓶中加入7.1g化合物v,加入30ml乙酸丁酯溶解,然后滴加15ml氯化氢-乙酸丁酯溶液(4m),25℃搅拌反应过夜。滴加饱和碳酸钠溶液调节ph至8,加入乙酸丁酯/水萃取,有机相水洗后减压浓缩干,得5.0g黄色油状物vi,收率85%。
34.(3)向250ml反应瓶中依次加入100ml四氢呋喃、5.0g化合物vi、2.8g化合物vii和1.2g的2,6-二甲基吡啶,60℃保温反应30小时。减压浓缩反应液,加水/乙酸乙酯萃取,水洗有机相,浓缩有机相,反应柱层析纯化,二氯甲烷∶甲醇体积比40∶1为洗脱液,收集目标组分后浓缩干,得到4.1g类白色固体viii,收率61%。
35.(4)向100ml反应瓶中依次加入30ml水、1.3g氢氧化钠,搅拌溶清后加入4.1g化合物viii,25℃反应2小时,缓慢滴加稀盐酸,调节ph至2~3,析出固体,抽滤,滤饼真空烘干,得到3.2g淡黄色固体雷替曲塞杂质i,收率90%。
36.实施例3(1)向250ml反应瓶中依次加入100ml乙腈、5g化合物ii、2.2g的hopo和3.7g的edci,45℃搅拌反应25分钟,而后加入3.4g化合物iii、2.4g三乙胺,45℃搅拌反应2.5小时。然后向所得反应液中再次加入2.2g的hopo和3.7g的edci,45℃搅拌反应30分钟,继续加入2.8g化合物iv、2.4g三乙胺,45℃搅拌反应3小时。减压浓缩反应液,加入二氯甲烷/水萃取,有机相水洗后减压浓缩干得到8.3g黄色油状物v,收率72%。
37.(2)向100ml反应瓶中加入8.3g化合物v,加入30ml无水乙醇溶解,然后滴加14ml氯化氢-乙醇溶液(5m),25℃搅拌反应过夜。将反应液冷至10℃以下,滴加饱和碳酸氢钠溶液调节ph至7.5,加入乙酸乙酯/水萃取,有机相水洗后减压浓缩干,得5.4g黄色油状物vi,收率78%。
38.(3)向250ml反应瓶中依次加入100ml2-甲基四氢呋喃、5.4g化合物vi、3.0g化合物vii和1.3g的2,6-二甲基吡啶,60℃保温反应36小时。减压浓缩反应液,加水/乙酸乙酯萃
取,水洗有机相,浓缩有机相,反应柱层析纯化,二氯甲烷∶甲醇体积比40∶1为洗脱液,收集目标组分后浓缩干,得到4.4g类白色固体viii,收率61%。
39.(4)向100ml反应瓶中依次加入25ml水、1.4g氢氧化钾,搅拌溶清后加入4.4g化合物viii,30℃反应2小时,缓慢滴加稀盐酸,调节ph至2~3,析出固体,抽滤,滤饼真空烘干,得到3.3g淡黄色固体雷替曲塞杂质i,收率87%。
40.本发明中所用原料、设备,若无特别说明,均为本领域的常用原料、设备;本发明中所用方法,若无特别说明,均为本领域的常规方法。
41.以上所述,仅是本发明的较佳实施例,并非对本发明作任何限制,凡是根据本发明技术实质对以上实施例所作的任何简单修改、变更以及等效变换,均仍属于本发明技术方案的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1