一种1H-咪唑类化合物的合成方法
一种1h-咪唑类化合物的合成方法
【技术领域】
1.本发明涉及有机合成领域,具体涉及一种由芳基甲基酮类化合物和胺类化合物合成1h-咪唑类化合物的方法。
背景技术:
2.咪唑类化合物是一种非常重要的含氮五元杂环化合物,其具有显著的生物医药活性。例如:eprosartan是一种非肽型血管紧张素ii受体拮抗剂,于2014年被美国食品药品监督管理局(fda)批准用于治疗高血压。咪唑的药物化学研究应用还包括hsp70的atp酶活性抑制剂apoptozole,消化性溃疡治疗剂cimetidine和质子泵抑制剂omeprazole等等。同时,该类化合物也可以作为一类重要的配体,离子液体,发光功能材料以及其它有用的有机中间体。
3.基于这一重要性,研究多取代1h-咪唑类化合物的合成具有重要意义。合成咪唑的传统方法包括以下策略:(1)1,2-二羰基化合物,氨和醛的三组分缩合,(2)α-二酮或者α-羟基酮与甲酰胺缩合环化。尽管取得了一些进展,但这些反应通常需要制备进行环化反应所需的中间体。此外,在某些情况下,昂贵且有毒的金属催化剂、非商业可用的起始材料、高反应温度或化学计量的强酸或强碱严重限制了其发展,寻找原子和步骤经济性的新策略仍然是科学家的极大兴趣。
4.芳基甲基酮是大多数反应中理想的合成子,较容易通过经典的friedel-crafts酰化和wacker型反应制备。据在先前的工作中,尽管一些以铜为催化剂,芳基甲基酮为底物的有效方法已被成功开发;然而,过渡金属的使用有时是有毒且昂贵的,可能在产品中残留痕量金属。简单、经济、有效的无金属氧化方法可以很好的避免这一问题,为大大扩展咪唑类化合物的应用提供可能。【参考文献:(a)j.yang,y.xu,y.yan,w.li,l.zhao,q.dai,y.li,s.li,j.zhong,r.cao and w.zhong,acs infect.dis.,2020,6,832;(b)j.h.tu,s.l.sheu and j.m.teng,jama dermatol,2016,152,1258;(c)k.j.m.and j.a.balfour,drugs,1998,55,713;(d)u.h.jin,s.k.michelhaugh,l.a.polin,r.shrestha,s.mittal and s.safe,cancers(basel),2020,12,2097.(e)y.yuan,j.-x.chen,f.lu,q.-x.tong,q.-d.yang,h.-w.mo,t.-w.ng,f.-l.wong,z.-q.guo,j.ye,z.chen,x.-h.zhang and c.-s.lee,chem.mater.,2013,25,4957;(f)j.dupont,r.f.de souza and p.a.z.suarez,chem.rev.,2002,102,3667.(g)p.l.arnold,s.t.liddle,j.mcmaster,c.jones and d.p.mills.j.am.chem.soc.,2007,129,5360;(h)b.radzisewski,eur.j.org.chem.,1882,15,2706;(i)h.bredreck,chem.ber.,1953,86,88.(j)p.k.s.kumari shalini,nitin kumar,2010,der chemica sinica,2010,1,36;(k)w.qian,l.zhang,h.sun,h.jiang and h.liu,adv.synth.catal.,2012,354,3231;(l)s.r-daryasarei,m.h.gohari and n.mohammadi,new j.chem.,2021,45,20486;(m)s.-y.w.z.-j.cai,s.-y.wang,and s.-j.ji,org.lett.,2012,14,6068;(n)h.huang,x.ji,w.wu and h.jiang,adv.synth.catal.,2013,355,170】
5.基于这一背景,开发一种以简单易得的芳基甲基酮类化合物为原料合成1h-咪唑类化合物的有效方法,对于含1h-咪唑骨架药物和材料的合成及应用研究具有非常重要的学术意义和工业应用价值。
技术实现要素:
6.本发明的目的是开发一种在氮气氛围下,以芳基甲基酮类化合物和胺类化合物为原料,在无金属条件下,在氧化剂作用下合成多取代1h-咪唑类化合物的方法。
7.本发明的目的是通过如下技术方案实现的:
8.一种1h-咪唑类化合物的合成方法,其特征在于,所述1h-咪唑类化合物的制备原料包括:芳基甲基酮类化合物、胺类化合物、氧化剂、2,2,6,6-四甲基哌啶氧化物(tempo)、酸;包含以下步骤:取芳基甲基酮类化合物、胺类化合物、氧化剂、tempo、酸、溶剂置于反应容器中,混合,搅拌反应,反应结束后冷却至室温,用饱和na2co3洗涤,然后用乙酸乙酯萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离,即得1h-咪唑类化合物;
9.所述1h-咪唑类化合物具有以下反应式:
[0010][0011]
所述反应式中,r选自苯基、4-甲基苯基、4-甲氧基苯基、2-氟苯基、4-氯苯基、4-硝基苯基、4-甲基磺酰基苯基、吡啶基、噻吩基中的一种;r1选自苯基、4-甲基苯基、4-叔丁基苯基、4-甲氧基苯基、4-氟苯基、4-氯苯基、3-氯苯基、2-氯苯基、4-溴苯基、4-碘苯基、4-三氟甲基苯基、萘基、呋喃基、噻吩基、吡啶基、叔丁基中的一种;
[0012]
所述氧化剂选自高碘酸钠、过硫酸钾、碘、过硫酸铵中的一种;
[0013]
所述酸选自冰醋酸、亚磷酸、三氟乙酸、硼酸中的一种;
[0014]
所述芳基甲基酮类化合物、胺类化合物、氧化剂、tempo、酸之间的摩尔比为1:(3.9~4.1):(2~2.2):(1.9~2.1):(1.9~2.1);
[0015]
所述芳基甲基酮类化合物选自于苯乙酮、对甲基苯乙酮、对甲氧基苯乙酮、2-氟苯乙酮、对氯苯乙酮、对硝基苯乙酮、对甲基磺酰基苯乙酮、1-(3-吡啶基)乙酮、2-噻吩乙酮中的一种;
[0016]
所述胺类化合物选自苯甲胺、对甲基苯甲胺、对叔丁基苯甲胺、对甲氧基苯甲胺、对氟苯甲胺、对氯苯甲胺、3-氯苯甲胺、2-氯苯甲胺、对溴苯甲胺、对碘苯甲胺、对三氟甲基苯甲胺、1-萘甲胺、2-呋喃甲胺、2-噻吩甲胺、3-吡啶基甲胺、叔戊胺的一种。
[0017]
优选的,一种由芳基甲基酮类化合物胺类化合物合成1h-咪唑类化合物的方法,包含以下步骤:取芳基甲基酮类化合物、胺类化合物、氧化剂、tempo、酸,混合;在惰性气体氛围下,加入溶剂,反应,得到咪唑类化合物。
[0018]
进一步优选的,一种由芳基甲基酮类化合物和胺类化合物合成1h-咪唑类化合物的方法,包含以下步骤:取芳基甲基酮类化合物、胺类化合物、氧化剂、tempo、酸,混合;在惰性气体氛围下,加入溶剂,搅拌反应,反应结束后冷却至室温,用饱和na2co3洗涤,萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离,即得1h-咪唑类化合物。
[0019]
更优选的,一种由芳基甲基酮类化合物和胺类化合物合成咪唑类化合物的方法,包含以下步骤:取芳基甲基酮类化合物、胺类化合物、氧化剂、tempo、酸,混合;在惰性气体氛围下,加入溶剂,搅拌反应,反应结束后冷却至室温,用饱和na2co3洗涤,然后用乙酸乙酯萃取,无水na2so4干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离,即得1h-咪唑类化合物。
[0020]
所述反应过程中氧化剂、tempo、酸的用量均为2当量。
[0021]
所述溶剂为1,2-二氯乙烷、乙腈、氯苯、甲苯、环己烷中的一种。
[0022]
所述反应的温度为50~90℃。
[0023]
优选的,所述反应的温度为70℃。
[0024]
所述反应的时间为6~24h。
[0025]
优选的,所述反应的时间为12h。
[0026]
所述惰性气体为氮气、氩气与氦气中的任意一种或多种的组合。
[0027]
根据实验研究,本发明提供了一种由芳基甲基酮类化合物和胺类化合物合成1h-咪唑类化合物的方法。该方法具有原料廉价易得、反应体系简单、所得目标产物易分离、反应操作简便、安全可靠等特点。该方法主要解决了1h-咪唑类化合物合成中副产物多、金属残留的难题,具体表现为:以简单易得而又容易制备的芳基甲基酮类化合物代替传统的α-羟基酮或α-二酮化合物为底物,避免了原料的预官能团化。该反应底物范围广,官能团耐受性较高,可用于合成一系列1,2,4-多取代的1h-咪唑类衍生物;同时,三氟甲基、硝基、卤素这些官能团取代的1h-咪唑也能在该体系中获得。
[0028]
【附图简要说明】
[0029]
图1为合成1h-咪唑类化合物的反应式。
【具体实施方式】
[0030]
下面结合本发明的合成例对本发明所述的合成方法作进一步说明,需要说明的是,实施例并不构成对本发明要求保护范围的限制。
[0031]
一种1h-咪唑类化合物的合成方法,其特征在于,所述1h-咪唑类化合物的制备原料包括:芳基甲基酮类化合物、胺类化合物、氧化剂、2,2,6,6-四甲基哌啶氧化物(tempo)、酸;包含以下步骤:取芳基甲基酮类化合物、胺类化合物、氧化剂、tempo、酸、溶剂置于反应容器中,混合,搅拌反应,反应结束后冷却至室温,用饱和na2co3洗涤,然后用乙酸乙酯萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离,即得1h-咪唑类化合物;
[0032]
所述1h-咪唑类化合物具有以下反应式:
[0033][0034]
所述反应式中,r选自苯基、4-甲基苯基、4-甲氧基苯基、2-氟苯基、4-氯苯基、4-硝基苯基、4-甲基磺酰基苯基、吡啶基、噻吩基中的一种;r1选自苯基、4-甲基苯基、4-叔丁基苯基、4-甲氧基苯基、4-氟苯基、4-氯苯基、3-氯苯基、2-氯苯基、4-溴苯基、4-碘苯基、4-三氟甲基苯基、萘基、呋喃基、噻吩基、吡啶基、叔丁基中的一种;
[0035]
所述芳基甲基酮类化合物、胺类化合物、氧化剂、tempo、酸之间的摩尔比为1:(3.9~4.1):(2~2.2):(1.9~2.1):(1.9~2.1)。
[0036]
在一种实施方式中,所述芳基甲基酮类化合物选自苯乙酮、对甲基苯乙酮、对甲氧基苯乙酮、2-氟苯乙酮、对氯苯乙酮、对硝基苯乙酮、对甲基磺酰基苯乙酮、1-(3-吡啶基)乙酮、2-噻吩乙酮中的一种。
[0037]
在一种实施方式中,所述胺类化合物选自苯甲胺、对甲基苯甲胺、对叔丁基苯甲胺、对甲氧基苯甲胺、对氟苯甲胺、对氯苯甲胺、3-氯苯甲胺、2-氯苯甲胺、对溴苯甲胺、对碘苯甲胺、对三氟甲基苯甲胺、1-萘甲胺、2-呋喃甲胺、2-噻吩甲胺、3-吡啶基甲胺、叔戊胺的一种。
[0038]
在一种实施方式中,一种由芳基甲基酮类化合物和胺类化合物合成咪唑类化合物的方法,包含以下步骤:取芳基甲基酮类化合物、胺类化合物、氧化剂、tempo、酸,混合;在惰性气体氛围下,加入溶剂,反应,得到1h-咪唑类化合物。
[0039]
在一种实施方式中,一种由芳基甲基酮类化合物和胺类化合物合成咪唑类化合物的方法,包含以下步骤:取芳基甲基酮类化合物、胺类化合物、氧化剂、tempo、酸,混合;在惰性气体氛围下,加入溶剂,搅拌反应,反应结束后冷却至室温,用饱和na2co3洗涤,萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离,即得1h-咪唑类化合物。
[0040]
在一种实施方式中,一种由芳基甲基酮类化合物和胺类化合物合成咪唑类化合物的方法,包含以下步骤:取芳基甲基酮类化合物、胺类化合物、氧化剂、tempo、酸,混合;在惰性气体氛围下,加入溶剂,搅拌反应,反应结束后冷却至室温,用饱和na2co3洗涤,然后用乙酸乙酯萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离,即得1h-咪唑类化合物。
[0041]
在一种实施方式中,所述溶剂为1,2-二氯乙烷、乙腈、氯苯、甲苯、环己烷中的一种。
[0042]
在一种实施方式中,所述反应的温度为50~90℃。
[0043]
在一种实施方式中,所述反应的温度为70℃。
[0044]
在一种实施方式中,所述反应的时间为6~24h。
[0045]
在一种实施方式中,所述反应的时间为12h。
[0046]
在一种实施方式中,所述惰性气体为氮气、氩气与氦气中的任意一种或多种的组合。
[0047]
根据实验研究,本发明提供了一种由芳基甲基酮类化合物和胺类化合物合成1h-咪唑类化合物的方法。该方法具有原料廉价易得、反应体系简单、所得目标产物易分离、反应操作简便、安全可靠等特点。该方法主要解决了1h-咪唑类化合物合成中副产物多、金属残留的难题,具体表现为:以简单易得而又容易制备的芳基甲基酮类化合物代替传统的α-羟基酮或α-二酮化合物为底物,避免了原料的预官能团化。该反应底物范围广,官能团耐受性较高,可用于合成一系列1,2,4-多取代的1h-咪唑类衍生物;同时,三氟甲基、硝基、卤素这些官能团取代的1h-咪唑也能在该体系中获得。
[0048]
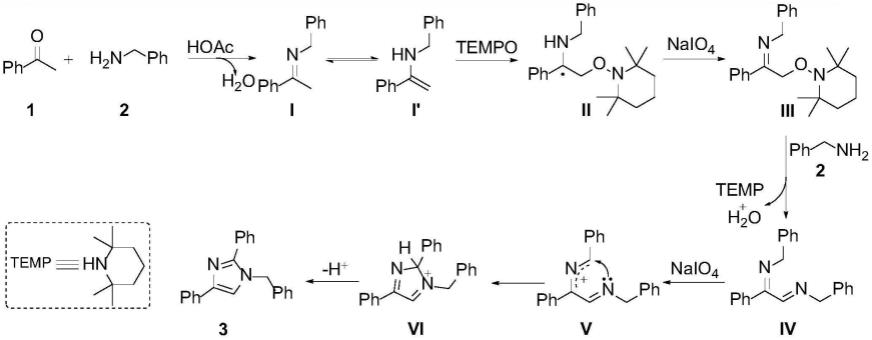
本发明通过以下反应途径获得(以苯乙酮和苯甲胺为例):
[0049][0050]
下面是具体的合成例。
[0051]
如图1所示,
[0052]
合成例1
[0053]
1-苄基-2,4-二苯基-1h-咪唑的合成
[0054]
在反应器中加入0.2mmol苯乙酮、0.8mmol苯甲胺、0.4mmol naio4、0.4mmol tempo、0.4mmol冰醋酸,2.0ml ch3cn。在氮气氛围下,在70℃条件下持续搅拌12h,停止反应,冷却至室温,用饱和nahco3洗涤,然后用乙酸乙酯萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离即得目标产物,产率88%。1h nmr(400mhz,cdcl3):δ7.88(d,j=7.6hz,2h),7.65(d,j=4.9hz,2h),7.46(d,j=4.6hz,3h),7.39(m,5h),7.27(d,j=10.4hz,2h),7.17(d,j=7.1hz,2h),5.24(s,2h).
13
c nmr(101mhz,cdcl3):δ147.5,140.4,135.7,133.0,129.3,127.9,127.7,127.5,127.4,126.9,125.7,125.6,123.8,115.7,49.4.
[0055]
合成例2
[0056]
1-(4-甲基苄基)-4-苯基-2-(对甲苯基)-1h-咪唑的合成
[0057]
在反应器中加入0.2mmol苯乙酮、0.8mmol 4-甲基苯甲胺、0.4mmol naio4、0.4mmol tempo、0.4mmol冰醋酸,2.0ml ch3cn。在氮气氛围下,在70℃条件下持续搅拌12h,停止反应,冷却至室温,用饱和nahco3洗涤,然后用乙酸乙酯萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离即得目标产物,产率91%。1h nmr(400mhz,cdcl3):δ7.92(d,j=7.4hz,2h),7.59(d,j=8.0hz,2h),7.44(t,j=7.7hz,2h),7.34
–
7.29(m,3h),7.28(s,1h),7.23(d,j=7.9hz,2h),7.10(d,j=8.0hz,2h),5.21(s,2h),2.46(s,3h),2.42(s,3h).
13
c nmr(101mhz,cdcl3):δ148.4,141.1,138.6,137.4,134.0,133.7,129.4,129.1,128.7,127.4,126.5,126.3,124.7,116.4,50.0,21.1,20.9.
[0058]
合成例3
[0059]
1-(4-(叔丁基)苄基)-2-(4-(叔丁基)苯基)-4-苯基-1h-咪唑的合成
[0060]
在反应器中加入0.2mmol苯乙酮、0.8mmol 4-叔丁基苯甲胺、0.4mmol naio4、0.4mmol tempo、0.4mmol冰醋酸,2.0ml ch3cn。在氮气氛围下,在70℃条件下持续搅拌12h,停止反应,冷却至室温,用饱和nahco3洗涤,然后用乙酸乙酯萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离即得目标产物,产率86%。1h nmr(400mhz,cdcl3):δ7.83(d,j=7.2hz,2h),7.56(d,j=7.5hz,2h),7.43(d,j=7.5hz,2h),7.34(t,j=7.8hz,4h),7.21(s,2h),7.07(d,j=7.4hz,2h),5.18(s,2h),1.33(s,9h),1.31(s,9h).
13
c nmr(101mhz,cdcl3):δ151.4,150.3,148.1,140.8,133.6,133.4,128.1,127.9,127.0,126.0,125.8,
125.3,125.0,124.3,116.0,49.5,34.1,33.9,30.7,30.6.
[0061]
合成例4
[0062]
1-(4-甲氧基苄基)-2-(4-甲氧基苯基)-4-苯基-1h-咪唑的合成
[0063]
在反应器中加入0.2mmol苯乙酮、0.8mmol 4-甲氧基苯甲胺、0.4mmol naio4、0.4mmol tempo、0.4mmol冰醋酸,2.0ml ch3cn。在氮气氛围下,在70℃条件下持续搅拌12h,停止反应,冷却至室温,用饱和nahco3洗涤,然后用乙酸乙酯萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离即得目标产物,产率41%。1h nmr(400mhz,cdcl3):δ7.84(d,j=7.6hz,2h),7.55(d,j=7.9hz,2h),7.36(t,j=7.3hz,2h),7.23(d,j=7.2hz,1h),7.19(d,j=6.4hz,1h),7.04(d,j=8.1hz,2h),6.95(d,j=8.0hz,2h),6.86(d,j=8.1hz,2h),5.08(s,2h),3.81(s,3h),3.77(s,3h).
13
c nmr(101mhz,cdcl3):δ160.0,159.1,148.2,140.9,134.1,130.2,128.7,128.3,127.9,126.5,124.7,122.9,116.3,114.2,113.9,55.1,54.9,49.8.
[0064]
合成例5
[0065]
1-(4-氟苄基)-2-(4-氟苯基)-4-苯基-1h-咪唑的合成
[0066]
在反应器中加入0.2mmol苯乙酮、0.8mmol 4-氟苯甲胺、0.4mmol naio4、0.4mmol tempo、0.4mmol冰醋酸,2.0ml ch3cn。在氮气氛围下,在70℃条件下持续搅拌12h,停止反应,冷却至室温,用饱和nahco3洗涤,然后用乙酸乙酯萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离即得目标产物,产率82%。1h nmr(400mhz,cdcl3):δ7.83(d,j=7.5hz,2h),7.66
–
7.51(m,2h),7.38(t,j=7.4hz,2h),7.29
–
7.21(m,2h),7.09(m,6h),5.16(s,2h).
13
c nmr(101mhz,cdcl3):δ164.0(d,j=80.8hz),161.6(d,j=78.9hz),147.5,141.6,133.8,132.3(d,j=3.3hz),130.9(d,j=8.4hz),128.6,128.4,128.3,126.9,116.7,116.1,115.9(d,j=6.0hz),115.6.
19
f nmr(376mhz,cdcl3)δ-111.6,-113.8.
[0067]
合成例6
[0068]
1-(3-氯苄基)-2-(3-氯苯基)-4-苯基-1h-咪唑的合成
[0069]
在反应器中加入0.2mmol苯乙酮、0.8mmol 3-氯苯甲胺、0.4mmol naio4、0.4mmol tempo、0.4mmol冰醋酸,2.0ml ch3cn。在氮气氛围下,在70℃条件下持续搅拌12h,停止反应,冷却至室温,用饱和nahco3洗涤,然后用乙酸乙酯萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离即得目标产物,产率87%。1h nmr(400mhz,cdcl3):δ7.81(d,j=7.5hz,2h),7.61(s,1h),7.36(q,j=6.4,5.5hz,4h),7.31(d,j=7.7hz,1h),7.24(d,j=9.8hz,3h),7.20(s,1h),7.07(s,1h),6.93(d,j=6.6hz,1h),5.11(s,2h).
13
c nmr(101mhz,cdcl3):δ146.8,141.8,138.4,134.9,134.6,133.6,131.8,130.3,129.8,129.1,129.0,128.5,128.3,127.0,126.7,126.6,124.8,124.6,117.1,49.9.合成例7
[0070]
1-(4-溴苄基)-2-(4-溴苯基)-4-苯基-1h-咪唑的合成
[0071]
在反应器中加入0.2mmol苯乙酮、0.8mmol 4-溴苯甲胺、0.4mmol naio4、0.4mmol tempo、0.4mmol冰醋酸,2.0ml ch3cn。在氮气氛围下,在70℃条件下持续搅拌12h,停止反应,冷却至室温,用饱和nahco3洗涤,然后用乙酸乙酯萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离即得目标产物,产率76%。1h nmr(400mhz,cdcl3):δ7.80(d,j=7.5hz,2h),7.53(d,j=7.9hz,2h),7.43(t,j=9.9hz,4h),7.35(t,j=7.4hz,2h),7.24(d,j=6.9hz,1h),7.19(s,1h),6.94(d,j=7.8hz,2h),5.09(s,2h).
13
c nmr(101mhz,cdcl3):δ
147.2,141.8,135.4,133.6,132.1,131.8,130.2,129.0,128.5,128.1,126.9,124.8,123.3,122.0,116.9,49.8.
[0072]
合成例8
[0073]
1-(4-碘苄基)-2-(4-碘苯基)-4-苯基-1h-咪唑的合成
[0074]
在反应器中加入0.2mmol苯乙酮、0.8mmol 4-碘苯甲胺、0.4mmol naio4、0.4mmol tempo、0.4mmol冰醋酸,2.0ml ch3cn。在氮气氛围下,在70℃条件下持续搅拌12h,停止反应,冷却至室温,用饱和nahco3洗涤,然后用乙酸乙酯萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离即得目标产物,产率62%。1h nmr(400mhz,cdcl3):δ7.79(d,j=7.6hz,2h),7.73(d,j=7.6hz,2h),7.64(d,j=7.6hz,2h),7.35(t,j=7.3hz,2h),7.28(d,j=7.6hz,2h),7.24(d,j=5.6hz,1h),7.19(s,1h),6.81(d,j=7.7hz,2h),5.08(s,2h).
13
c nmr(101mhz,cdcl3):δ147.3,141.8,138.0,137.7,136.1,133.6,129.6,128.5,128.3,127.0,124.8,117.0,95.2,93.5,49.9.
[0075]
合成例9
[0076]
4-苯基-1-(4-(三氟甲基)苄基)-2-(4-(三氟甲基)苯基)-1h-咪唑的合成
[0077]
在反应器中加入0.2mmol苯乙酮、0.8mmol 4-三氟甲基苯甲胺、0.4mmol naio4、0.4mmol tempo、0.4mmol冰醋酸,2.0ml ch3cn。在氮气氛围下,在70℃条件下持续搅拌12h,停止反应,冷却至室温,用饱和nahco3洗涤,然后用乙酸乙酯萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离即得目标产物,产率78%。1h nmr(400mhz,cdcl3):δ7.85(d,j=7.6hz,2h),7.77
–
7.66(m,4h),7.63(d,j=7.8hz,2h),7.39(t,j=7.3hz,2h),7.28(s,2h),7.26
–
7.12(m,2h),5.29(s,2h).
13
c nmr(101mhz,cdcl3):δ147.0,142.5,140.4,133.6,133.5,131.1(d,j=9.9hz),130.8(d,j=9.9hz),130.5(d,j=9.8hz),129.1,128.7,127.3,126.7,126.2(q,j=3.6hz),125.7(q,j=3.6hz),125.0,122.5(d,j=7.5hz),117.4,50.2.
19
f nmr(376mhz,cdcl3):δ-62.7(s,1f),-62.8(s,1f).
[0078]
合成例10
[0079]
2-(萘-1-基)-1-(萘-1-基甲基)-4-苯基-1h-咪唑的合成
[0080]
在反应器中加入0.2mmol苯乙酮、0.8mmol萘甲胺、0.4mmol naio4、0.4mmol tempo、0.4mmol冰醋酸,2.0ml ch3cn。在氮气氛围下,在70℃条件下持续搅拌12h,停止反应,冷却至室温,用饱和nahco3洗涤,然后用乙酸乙酯萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离即得目标产物,产率79%。1h nmr(400mhz,cdcl3):δ7.94(q,j=12.7,10.2hz,3h),7.84(d,j=7.9hz,3h),7.80(d,j=8.5hz,1h),7.65(d,j=7.0hz,1h),7.61(d,j=8.4hz,1h),7.57
–
7.52(m,2h),7.51(d,j=8.2hz,1h),7.46(d,j=7.9hz,1h),7.43(d,j=5.6hz,1h),7.39(d,j=9.4hz,1h),7.34(t,j=6.7hz,3h),7.27(s,1h),7.20(d,j=7.0hz,1h),5.40(s,2h).
13
c nmr(101mhz,cdcl3):δ147.0,141.5,134.1,133.8,133.7,132.9,131.9,130.6,130.0,128.9,128.8,128.7,128.5,128.3,128.0,127.0,126.7,126.6,126.3,126.1,125.9,125.8,125.4,125.0,124.9,122.5,115.8,48.6.
[0081]
合成例11
[0082]
1-苄基-4-(4-硝基苯基)-2-苯基-1h-咪唑的合成
[0083]
在反应器中加入0.2mmol 4-硝基苯乙酮、0.8mmol苯甲胺、0.4mmol naio4、0.4mmol tempo、0.4mmol冰醋酸,2.0ml ch3cn。在氮气氛围下,在70℃条件下持续搅拌12h,
停止反应,冷却至室温,用饱和nahco3洗涤,然后用乙酸乙酯萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离即得目标产物,产率91%。1h nmr(400mhz,cdcl3):δ8.22(d,j=7.7hz,2h),7.96(d,j=7.8hz,2h),7.66
–
7.59(m,2h),7.46(s,3h),7.41(s,1h),7.37(d,j=7.8hz,2h),7.26(s,1h),7.15(d,j=7.0hz,2h),5.25(s,2h).
13
c nmr(101mhz,cdcl3):δ149.6,146.3,140.5,139.4,136.2,129.9,129.5,129.2,129.0,128.8,128.3,126.8,125.1,124.1,119.1,50.8.
[0084]
合成例12
[0085]
1-苄基-4-(4-(甲基磺酰基)苯基)-2-苯基-1h-咪唑的合成
[0086]
在反应器中加入0.2mmol 4-甲基磺酰基基苯乙酮、0.8mmol苯甲胺、0.4mmol naio4、0.4mmol tempo、0.4mmol冰醋酸,2.0ml ch3cn。在氮气氛围下,在70℃条件下持续搅拌12h,停止反应,冷却至室温,用饱和nahco3洗涤,然后用乙酸乙酯萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离即得目标产物,产率88%。1h nmr(400mhz,cdcl3):δ8.01(d,j=7.7hz,2h),7.91(d,j=7.9hz,2h),7.64
–
7.57(m,2h),7.45(s,3h),7.37(d,j=12.9hz,3h),7.26(s,1h),7.14(d,j=7.0hz,2h),5.24(s,2h),3.05(s,3h).
13
c nmr(101mhz,cdcl3):δ149.4,139.6,139.5,138.0,136.3,130.0,129.4,129.1,129.0,128.7,128.2,127.7,126.8,125.3,118.7,50.7,44.6.
[0087]
合成例13
[0088]
1-苄基-2-苯基-4-(吡啶-3-基)-1h-咪唑的合成
[0089]
在反应器中加入0.2mmol 1-(3-吡啶基)乙酮、0.8mmol苯甲胺、0.4mmol naio4、0.4mmol tempo、0.4mmol冰醋酸,2.0ml ch3cn。在氮气氛围下,在70℃条件下持续搅拌12h,停止反应,冷却至室温,用饱和nahco3洗涤,然后用乙酸乙酯萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离即得目标产物,产率90%。1h nmr(400mhz,cdcl3):δ8.99(s,1h),8.44(d,j=3.5hz,1h),8.15(d,j=7.8hz,1h),7.63
–
7.51(m,2h),7.44
–
7.39(m,3h),7.31(m,5h),7.12(d,j=7.1hz,2h),5.22(s,2h).
13
c nmr(101mhz,cdcl3):δ149.2,147.7,146.4,138.4,136.5,132.2,130.0,129.2,129.1,129.0,128.7,128.1,127.8,127.5,126.7,117.3,50.6.
[0090]
合成例14
[0091]
1-苄基-2-苯基-4-(噻吩-2-基)-1h-咪唑的合成
[0092]
在反应器中加入0.2mmol 1-(噻吩-2-基)乙烷-1-酮、0.8mmol苯甲胺、0.4mmol naio4、0.4mmol tempo、0.4mmol冰醋酸,2.0ml ch3cn。在氮气氛围下,在70℃条件下持续搅拌12h,停止反应,冷却至室温,用饱和nahco3洗涤,然后用乙酸乙酯萃取,干燥,减压蒸馏浓缩除去溶剂,粗产品经柱色谱分离即得目标产物,产率74%。1h nmr(400mhz,cdcl3):δ7.62
–
7.57(m,2h),7.43
–
7.39(m,3h),7.39
–
7.31(m,4h),7.18(d,j=4.6hz,1h),7.13(d,j=9.1hz,3h),7.03(s,1h),5.18(s,2h).
13
c nmr(101mhz,cdcl3):δ148.4,137.8,136.7,136.6,130.1,129.0,128.9,128.5,127.9,127.4,126.6,124.9,123.2,122.0,116.2,50.4.
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1