一种醛酮类化合物的合成工艺
1.本发明涉及有机中间体合成技术领域,尤其涉及一种醛酮类化合物的合成工艺。
背景技术:
2.醛酮类化合物是自然界中常见的物质,广泛存在于具有生物、药理活性的天然产物和人工合成的化合物中,在日常生产生活中可以用作香料、药物以及化工原料。该类物质可以发生几乎所有类型的有机转化,如氧化、还原、缩合、加成、环加成、偶联以及聚合反应等,可见醛酮类化合物在有机合成以及工业应用等领域具有不可替代的作用,因此该类的物质的高效合成具有重要的理论意义和实用价值。目前,醛酮类物质大多数由醇氧化直接得到,具体方法有:
3.方法一:以三氧化铬
·
吡啶的晶体为氧化剂,以二氯甲烷为溶剂,将醇氧化生成醛酮类物质。
[0004][0005]
该方法的弊端是需要使用过量(约6当量)的重金属氧化剂三氧化铬
·
吡啶,该反应原子利用率低,三废排放量大,严重污染环境。
[0006]
方法二:以四氯化碳作溶剂,先使用氯气和二甲基硫在0℃条件下生成硫盐中间体,该硫盐然后在三乙胺作为碱,温度为-25℃的条件下与醇反应生成醛酮类物质。
[0007][0008]
该方法的弊端在于反应所需的氯气、四氯化碳均具有毒性,容易对人体造成伤害,污染环境,使用也不便,所用硫盐不太稳定,只能原位生成并使用。
[0009]
方法三:在80℃条件下,以醋酸钯作催化剂,醇类化合物在氧气和二甲亚砜的共同作用下反应生成醛酮类物质。
[0010][0011]
该方法的弊端在于需要使用的重金属钯催化剂价格较为昂贵,回收困难,所用的二甲亚砜较难处理,反应所需时间较长。
[0012]
方法四:在室温和dmf作溶剂的条件下,醇类化合物受到催化剂tempo和助催化剂cucl 的共同作用,转化为醛酮类物质。
[0013][0014]
该反应的不足之处是使用dmf作溶剂,虽然对活泼的伯醇能有效地氧化,但对仲醇的氧化效果不佳,甚至不反应,底物范围的限制性很大。
技术实现要素:
[0015]
针对上述存在的问题,本发明目的在于提供一种成本低廉、操作简便、反应条件温和,易于在工业生产推广的醛酮类化合物的合成工艺。
[0016]
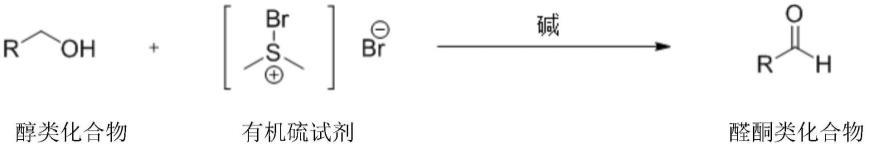
为了达到上述目的,本发明采用的技术方案如下:一种醛酮类化合物的合成工艺,所述工艺的合成方法如下:
[0017][0018]
其中,有机硫试剂为溴化二甲基溴化硫;碱为有机碱或无机碱;
[0019]
r为取代基,取代基选自:c
2-c
20
烯基、包含二级取代基的c
6-c
20
芳基、包含1-5个o、 n、s杂原子的5-10元杂芳基或共轭羰基;二级取代基选自:卤素、c
1-c
20
烷基或共轭羰基。
[0020]
本发明的取代基r优选包含二级取代基的c
6-c
20
芳基、包含1-5个o、n、s杂原子的 5-10元杂芳基,其中,二级取代基选自:卤素、c
1-c
20
烷基或苯环;当取代基r为芳基或5-10 元杂芳基时,通过本工艺生产出的产品收率最高。
[0021]
本发明所述的合成方法中有机硫试剂和醇类化合物的摩尔比为1~2:1;优选的摩尔比为 1.2:1或1.5:1;当有机硫试剂和醇类化合物的摩尔比为1.2:1或1.5:1时,最终产品的收率较高。
[0022]
本发明所述的合成方法中碱和有机硫试剂的摩尔比为1~2:1,优选的的摩尔比为2:1;当碱和有机硫试剂的摩尔比为2:1时,最终产品的收率达到最高。
[0023]
本发明所述的合成方法中反应溶剂为四氢呋喃或二氯甲烷;原料醇类化合物在反应溶剂中摩尔浓度为0.1mmol/ml;本发明在单一有机溶剂的体系中进行;如果需要,体系中也可以存在其它有机溶剂,但是从反应产率、操作的简洁性角度考虑,优选为不加其它有机溶剂,即以单一有机溶剂作为反应溶剂。
[0024]
本发明所述的合成方法中有机碱为一级胺、二级胺、三级胺、吡啶、dmap、dbu中的一种。本发明所述的合成方法中无机碱为碳酸钾、碳酸钠、碳酸铯、磷酸钾、磷酸钠中的一种;优选的碱为dbu。
[0025]
本发明所述的合成方法中反应温度为0℃~-78℃,优选-78℃;反应时间为15min~18h,优选30min。本发明的反应温度和反应时间可以由技术人员按照不同的醇类化合物,根据实际需要自行确定。
[0026]
本发明提供一种醛酮类化合物的精制方法,其精制方法如下:反应完成后,用水洗涤反应液并用乙酸乙酯萃取三次,经柱层析分离,得到精制后的醛酮类化合物。
[0027]
本发明的优点在于:本发明用简单易得的醇类化合物作为反应底物,用商业可购买、制作简单且对空气不易敏感的溴化二甲基溴化硫作为反应试剂,采用廉价易得的dbu作为碱,反应温度为-78℃逐渐恢复至室温,在氮气条件下简单而高效地合成了醛酮类化合物。与其它合成醛酮类化合物的方法相比,本发明的反应条件温和、所使用的的反应原料(包括醇、溴化二甲基溴化硫、dbu)均廉价易得、不需要使用金属催化剂、反应产率高,具有节约成本、环境友好、可工业推广的特点。
[0028]
本发明是一种适用范围广的醛酮合成的方法,对于芳香醛酮、脂肪醛酮、α,β-不饱
和醛酮、杂芳基醛酮的合成都具有良好的适用性。因此实际上对于醇类化合物及其衍生物中的取代基的个数和种类没有特别严格的限制,进而对于醛酮类化合物中的取代基的个数和种类也没有特别严格的限制。
[0029]
本发明对于多种醇类化合物均可适用,这也包括仲醇或烯丙醇,进而可以制得多种多样的醛酮类化合物;本发明可以广泛应用于工业和学术界的药物合成、天然产物的全合成中,具有较高的应用价值。
附图说明
[0030]
图1为实施例1所述对甲氧基苯甲醛的核磁氢谱图;
[0031]
图2为实施例1所述对甲氧基苯甲醛的核磁碳谱图;
[0032]
图3为实施例2所述3,5-二甲氧基苯甲醛的核磁氢谱图;
[0033]
图4为实施例3所述胡椒醛的核磁氢谱图;
[0034]
图5为实施例5所述4-联苯甲醛的核磁氢谱图;
[0035]
图6为实施例10所述1-萘甲醛的核磁氢谱图;
[0036]
图7为实施例11所述2-萘甲醛的核磁氢谱图;
[0037]
图8为实施例12所述9-蒽甲醛的核磁氢谱图;
[0038]
图9为实施例13所述二苯甲酮的核磁氢谱图;
[0039]
图10为实施例13所述二苯甲酮的核磁碳谱图;
[0040]
图11为实施例15所述9-芴酮的核磁氢谱图;
[0041]
图12为实施例15所述苯偶酰的核磁氢谱图。
具体实施方式
[0042]
下面结合附图说明和具体实施方式对本发明作进一步详细的描述。
[0043]
以下具体实施例中所用的原料均可商业购买,各试剂必要时采用本领域公知的手段进行纯化后使用。
[0044]
在本发明中,“醛酮类化合物”具有本领域技术人员所通常理解的含义,即含有羰基(-co-) 或醛基(-cho)的化合物,例如对甲氧基苯甲醛、肉桂醛、苯乙酮、二苯甲酮及其各种衍生物。
[0045]
在本发明中,“醇类化合物”具有本领域技术人员所通常理解的含义,即含有与羟基上的氧原子连接的烷基、烯基、苯基、杂环结构的化合物,例如对甲氧基苯甲醇、对溴苯甲醇、肉桂醇、2-吡啶甲醇、1-苯乙醇及其各种衍生物。
[0046]
以下具体实施例中所用的原料均可商业购买,各试剂必要时采用本领域公知的手段进行纯化后使用。
[0047]1h nmr和
13
c nmr均采用bruker avance 400spectrometer仪器进行测定。测试温度为室温,溶剂为氘代氯仿,选取参考:1h nmr:chcl3为7.260ppm;
13
c nmr:chcl3为77.000ppm。
[0048]
实施例1:对甲氧基苯甲醛的合成
[0049]
在氮气保护的schlenk反应管中加入对甲氧基苯甲醇(124.2mg,110μl)和dcm(4.5ml)溶解形成对甲氧基苯甲醇的溶液。另在装有磁力搅拌子的schlenk反应管中加入溴
nmr(101mhz,cdcl3)δ191.1,135.0,132.4,130.9,129.8ppm.
[0060]
实施例5:4-联苯甲醛的合成
[0061]
在氮气保护的schlenk反应管中加入4-联苯甲醇(124.6mg,110μl)和dcm(4.5ml),溶解形成4-联苯甲醇的溶液。另在装有磁力搅拌子的schlenk反应管中加入溴化二甲基溴化硫(100.0 mg,1.5equiv),在氮气保护氛围下放置于-78℃的低温反应槽中冷却10分钟,再于10分钟内逐滴加入4-联苯甲醇溶液(1.5ml,1.0equiv)和dbu(137.0mg,132μl,3.0equiv),滴加后搅拌溶液30分钟。反应完成后取出反应管恢复至室温,加入蒸馏水以淬灭反应,用乙酸乙酯进行萃取3次,每次10ml,合并有机相,经过旋蒸浓缩然后进行柱层析得到4-联苯甲醛27.3mg,产率为50%。
[0062]
产物4-联苯甲醛:1h nmr(400mhz,cdcl3)δ10.06(s,1h),8.02
–
7.89(m,2h),7.82
–ꢀ
7.72(m,2h),7.64(m,j=4.4,3.5,1.9hz,2h),7.54
–
7.38(m,3h)ppm.
13
c nmr(101mhz, cdcl3)δ191.9,147.1,139.6,135.1,130.2,129.0,128.4,127.6,127.3ppm.
[0063]
实施例6:对甲基苯甲醛的合成
[0064]
在氮气保护的schlenk反应管中加入对甲基苯甲醇(108.1mg)和dcm(4.5ml),溶解形成对甲基苯甲醇的溶液。另在装有磁力搅拌子的schlenk反应管中加入溴化二甲基溴化硫(100.0 mg,1.5equiv),在氮气保护氛围下放置于-78℃的低温反应槽中冷却10分钟,再于10分钟内逐滴加入对甲基苯甲醇溶液(1.5ml,1.0equiv)和dbu(137.0mg,132μl,3.0equiv),滴加后搅拌溶液30分钟。反应完成后取出反应管恢复至室温,加入蒸馏水以淬灭反应,用乙酸乙酯进行萃取3次,每次10ml,合并有机相,经过旋蒸浓缩然后进行柱层析得到对甲基苯甲醛15.2mg,产率为42%。
[0065]
产物对甲基苯甲醛:1h nmr(400mhz,cdcl3)δ9.96(s,1h),7.78(d,j=8.1hz,2h),7.33 (d,j=8.0hz,2h),2.44(s,3h)ppm.
13
c nmr(101mhz,cdcl3)δ192.0,145.5,134.1,129.8, 129.7,21.9ppm.
[0066]
实施例7:苯乙酮的合成
[0067]
在氮气保护的schlenk反应管中加入1-苯乙醇(123.1mg,123μl)和dcm(4.5ml),溶解形成1-苯乙醇的溶液。另在装有磁力搅拌子的schlenk反应管中加入溴化二甲基溴化硫(100.0mg, 1.5equiv),在氮气保护氛围下放置于-78℃的低温反应槽中冷却10分钟,再于10分钟内逐滴加入1-苯乙醇溶液(1.5ml,1.0equiv)和dbu(137.0mg,132μl,3.0equiv),滴加后搅拌溶液30分钟。反应完成后取出反应管恢复至室温,加入蒸馏水以淬灭反应,用乙酸乙酯进行萃取3次,每次10ml,合并有机相,经过旋蒸浓缩然后进行柱层析得到苯乙酮25.8mg,产率为64%。
[0068]
产物苯乙酮:1h nmr(400mhz,cdcl3)δ7.70
–
7.60(m,2h),7.28
–
7.20(m,1h),7.17
–ꢀ
7.05(m,2h),2.24(s,3h)ppm.
13
c nmr(101mhz,cdcl3)δ196.8,136.3,132.2,127.7,127.4, 25.6ppm.
[0069]
实施例8:2-吡啶甲醛的合成
[0070]
在氮气保护的schlenk反应管中加入2-吡啶甲醇(98.0mg,87μl)和dcm(4.5ml),溶解形成2-吡啶甲醇的溶液。另在装有磁力搅拌子的schlenk反应管中加入溴化二甲基溴化硫(100.0 mg,1.5equiv),在氮气保护氛围下放置于-78℃的低温反应槽中冷却10分钟,再于10分钟内逐滴加入2-吡啶甲醇溶液(1.5ml,1.0equiv)和dbu(137.0mg,132μl,
3.0equiv),滴加后搅拌溶液 30分钟。反应完成后取出反应管恢复至室温,加入蒸馏水以淬灭反应,用乙酸乙酯进行萃取3 次,每次10ml,合并有机相,经过旋蒸浓缩然后进行柱层析得到2-吡啶甲醛11.9mg,产率为37%。
[0071]
实施例9:2-噻吩甲醛的合成
[0072]
在氮气保护的schlenk反应管中加入2-噻吩甲醇(103.0mg,85μl)和dcm(4.5ml),溶解形成2-噻吩甲醇的溶液。另在装有磁力搅拌子的schlenk反应管中加入溴化二甲基溴化硫(100.0 mg,1.5equiv),在氮气保护氛围下放置于-78℃的低温反应槽中冷却10分钟,再于10mins内逐滴加入2-噻吩甲醇溶液(1.5ml,1.0equiv)和dbu(137.0mg,132μl,3.0equiv),滴加后搅拌溶液30分钟。反应完成后取出反应管恢复至室温,加入蒸馏水以淬灭反应,用乙酸乙酯进行萃取3次,每次10ml,合并有机相,经过旋蒸浓缩然后进行柱层析得到2-噻吩甲醛14.1mg,产率为42%。
[0073]
实施例10:1-萘甲醛的合成
[0074]
在氮气保护的schlenk反应管中加入1-萘甲醇(142.4mg)和dcm(4.5ml),溶解形成1-萘甲醇的溶液。另在装有磁力搅拌子的schlenk反应管中加入溴化二甲基溴化硫(100.0mg,1.5 equiv),在氮气保护氛围下放置于-78℃的低温反应槽中冷却10分钟,再于10分钟内逐滴加入 1-萘甲醇溶液(1.5ml,1.0equiv)和dbu(137.0mg,132μl,3.0equiv),滴加后搅拌溶液30分钟。反应完成后取出反应管恢复至室温,加入蒸馏水以淬灭反应,用乙酸乙酯进行萃取3次,每次 10ml,合并有机相,经过旋蒸浓缩然后进行柱层析得到1-萘甲醛30.0.mg,产率为64%。
[0075]
产物1-萘甲醛:1h nmr(400mhz,cdcl3)δ10.30(s,1h),9.24(m,j=8.6,0.6hz,1h), 7.96(d,j=8.2hz,1h),7.93
–
7.00(m,5h)ppm.
13
c nmr(101mhz,cdcl3)δ193.2,136.4, 134.9,133.3,130.9,130.1,128.7,128.2,126.6,124.5ppm.
[0076]
实施例11:2-萘甲醛的合成
[0077]
在氮气保护的schlenk反应管中加入2-萘甲醇(124.3mg)和dcm 4.5ml,溶解形成2-萘甲醇的溶液。另在装有磁力搅拌子的schlenk反应管中加入溴化二甲基溴化硫(100.0mg,1.5 equiv),在氮气保护氛围下放置于-78℃的低温反应槽中冷却10分钟,再于10分钟内逐滴加入 2-萘甲醇溶液(1.5ml,1.0equiv)和dbu(137.0mg,132μl,3.0equiv),滴加后搅拌溶液30分钟。反应完成后取出反应管恢复至室温,加入蒸馏水以淬灭反应,用乙酸乙酯进行萃取3次,每次 10ml,合并有机相,经过旋蒸浓缩然后进行柱层析得到2-萘甲醛35.6mg,产率为76%。
[0078]
产物2-萘甲醛:1h nmr(400mhz,cdcl3)δ10.16(s,1h),8.33(s,1h),8.04
–
7.86(m,4h), 7.62(m,j=16.2,6.9,1.3hz,2h)ppm.
13
c nmr(101mhz,cdcl3)δ192.2,136.4,134.5,134.0, 132.6,129.5,129.1,129.1,128.0,127.1,122.7ppm.
[0079]
实施例12:9-蒽甲醛的合成
[0080]
在氮气保护的schlenk反应管中加入9-蒽甲醇(187.4mg)和dcm(4.5ml),溶解形成9-蒽甲醇的溶液。另在装有磁力搅拌子的schlenk反应管中加入溴化二甲基溴化硫(100.0mg,1.5 equiv),在氮气保护氛围下放置于-78℃的低温反应槽中冷却10分钟,再于10分钟内逐滴加入 9-蒽甲醇溶液(1.5ml,1.0equiv)和dbu(137.0mg,132μl,3.0equiv),滴加后搅拌溶液30分钟。反应完成后取出反应管恢复至室温,加入蒸馏水以淬灭反应,用乙酸
乙酯进行萃取3次,每次 10ml,合并有机相,经过旋蒸浓缩然后进行柱层析得到9-蒽甲醛34.7mg,产率为56%。
[0081]
产物9-蒽甲醛:1h nmr(400mhz,cdcl3)δ11.52(s,1h),8.99(d,j=9.0hz,2h),8.69(s, 1h),8.06(d,j=8.4hz,2h),7.68(m,j=8.3,7.3hz,2h),7.62
–
7.48(m,2h)ppm.
13
c nmr (101mhz,cdcl3)δ193.0,135.3,132.2,131.1,129.3,129.2,125.7,123.6.
[0082]
实施例13:二苯甲酮的合成
[0083]
在氮气保护的schlenk反应管中加入二苯甲醇(165.8mg)和dcm(4.5ml),溶解形成二苯甲醇的溶液。另在装有磁力搅拌子的schlenk反应管中加入溴化二甲基溴化硫(100.0mg,1.5 equiv),在氮气保护氛围下放置于-78℃的低温反应槽中冷却10分钟,再于10分钟内逐滴加入二苯甲醇溶液(1.5ml,1.0equiv)和dbu(137.0mg,132μl,3.0equiv),滴加后搅拌溶液30分钟。反应完成后取出反应管恢复至室温,加入蒸馏水以淬灭反应,用乙酸乙酯进行萃取3次,每次10ml,合并有机相,经过旋蒸浓缩然后进行柱层析得到二苯甲酮50.8mg,产率为93%。
[0084]
产物二苯甲酮:1h nmr(400mhz,cdcl3)δ7.81(m,j=8.4,1.6hz,2h),7.63
–
7.54(m, 1h),7.53
–
7.43(m,2h)ppm.
13
c nmr(101mhz,cdcl3)δ196.7,137.5,132.3,130.0,128.2 ppm.
[0085]
实施例14:9-芴酮的合成
[0086]
在氮气保护的schlenk反应管中加入9-芴醇(162.2mg)和dcm(4.5ml),溶解形成9-芴醇的溶液。另在装有磁力搅拌子的schlenk反应管中加入溴化二甲基溴化硫(100.0mg,1.5equiv),在氮气保护氛围下放置于-78℃的低温反应槽中冷却10分钟,再于10分钟内逐滴加入9-芴醇溶液(1.5ml,1.0equiv)和dbu(137.0mg,132μl,3.0equiv),滴加后搅拌溶液30分钟。反应完成后取出反应管恢复至室温,加入蒸馏水以淬灭反应,用乙酸乙酯进行萃取3次,每次10ml,合并有机相,经过旋蒸浓缩然后进行柱层析得到9-芴酮43.3mg,产率为80%。
[0087]
产物9-芴酮:1h nmr(400mhz,cdcl3)δ7.62(d,j=7.4hz,1h),7.55
–
7.38(m,2h), 7.33
–
7.21(m,1h)ppm.
13
c nmr(101mhz,cdcl3)δ193.8,144.3,134.6,134.0,129.0,124.2, 120.2ppm.
[0088]
实施例15:苯偶酰的合成
[0089]
在氮气保护的schlenk反应管中加入安息香(191.8mg)和dcm(4.5ml),溶解形成安息香的溶液。另在装有磁力搅拌子的schlenk反应管中加入溴化二甲基溴化硫(100.0mg,1.5equiv),在氮气保护氛围下放置于-78℃的低温反应槽中冷却10分钟,再于10分钟内逐滴加入安息香溶液(1.5ml,1.0equiv)和dbu(137.0mg,132μl,3.0equiv),滴加后搅拌溶液30分钟。反应完成后取出反应管恢复至室温,加入蒸馏水以淬灭反应,用乙酸乙酯进行萃取3次,每次10ml,合并有机相,经过旋蒸浓缩然后进行柱层析得到苯偶酰56.8mg,产率为90%。
[0090]
产物苯偶酰:1h nmr(400mhz,cdcl3)δ8.00
–
7.95(m,4h),7.65(m,j=10.7,4.2hz, 2h),7.51(t,j=7.7hz,4h)ppm.
13
c nmr(101mhz,cdcl3)δ194.6,134.9,132.9,129.8,129.0 ppm.
[0091]
实施例16:肉桂醛的合成
[0092]
在氮气保护的schlenk反应管中加入肉桂醇(120.8mg,116μl)和dcm(4.5ml),溶解形成肉桂醇的溶液。另在装有磁力搅拌子的schlenk反应管中加入溴化二甲基溴化硫(100.0mg,1.5 equiv),在氮气保护氛围下放置于-78℃的低温反应槽中冷却10分钟,再于10分钟内逐滴加入肉桂醇溶液(1.5ml,1.0equiv)和dbu(137.0mg,132μl,3.0equiv),滴加后搅拌溶液30分钟。反应完成后取出反应管恢复至室温,加入蒸馏水以淬灭反应,用乙酸乙酯进行萃取3次,每次 10ml,合并有机相,经过旋蒸浓缩然后进行柱层析得到肉桂醛28.6mg,产率为72%。
[0093]
产物肉桂醛:1h nmr(400mhz,cdcl3)δ9.52
–
9.46(m,1h),7.36
–
7.31(m,2h),7.26
–ꢀ
7.18(m,4h),6.54
–
6.45(m,1h)ppm.
13
c nmr(101mhz,cdcl3)δ192.8,151.9,133.3,130.5, 128.4,127.8,127.7ppm.
[0094]
[0095][0096]
从实施例1-16可知,本发明的方法是从各类廉价易得的醇类化合物出发,以商业可购买的溴化二甲基溴化硫作为反应试剂,使用廉价易得的dbu作为有机碱,在氮气条件下于-78℃至室温时逐渐发生反应,得到所述的醛酮类化合物。该方法无论对于芳香醛酮、脂肪醛酮、α, β-不饱和醛酮、杂芳基醛酮以及含其他官能团或杂原子的醛酮类化合物均具有良好的适用性,是一种条件温和、操作简单的醛酮类化合物的通用合成方法。
[0097]
实施例17:对甲氧基苯甲醛的合成条件优化
[0098][0099]
标准条件:1 0.3mmol(1.0eq.),碱0.9mmol(3.0eq.),溶剂3.0ml,-78℃.分离产率.
[0100]
dbu=1,8-二氮杂双环[5.4.0]十一碳-7-烯;
[0101]
thf=四氢呋喃;
[0102]
dcm=二氯甲烷;
[0103]
dipa=二异丙醇胺;
[0104]
dipea=n,n-二异丙基乙基胺.
[0105]
需要说明的是,上述仅仅是本发明的较佳实施例,并非用来限定本发明的保护范围,在上述实施例的基础上所做出的任意组合或等同变换均属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1