一种外泌体分离富集方法及其应用
1.本发明属于生物技术领域,尤其涉及一种外泌体分离富集方法及其应用。
背景技术:
2.外泌体(exosome)是细胞向外分泌的小囊泡,直径多介于30~150nm之间,存在于各种体液中,如腹水、血液、脑脊液、泪液、尿液以及唾液等。外泌体携带大量胞内和胞外的生物信息,能够被受体细胞接收,实现细胞间的物质运输和信息传递,可作为细胞通讯和细胞外基质重塑的媒介,与人类健康和疾病息息相关。几乎所有的生物细胞在各种状态下都能释放外泌体,甚至包括一些微生物也会释放外泌体进行信号传递等功能。外泌体可以通mirna或dna等核酸进行信息传递,其也含有许多关键的蛋白质和特殊遗传物质,外泌体在细胞通讯和表观遗传调控中发挥着抗原呈递、免疫调节、组织发展、胞间通讯、介导肿瘤发生发展等重要作用。由于外泌体可以在体液(例如血液、尿液、唾液和脑脊髓液)中检测到,因此它们被认为是疾病诊断的无创或微创生物标志物,具有检测包括癌症在内的许多病理状况的潜力。现外泌体常应用于疾病的分子标志物、药物传输载体等。而外泌体可靠的分离与检测方法则是进行外泌体相关研究的重要前提。
3.脱氧核糖核酶(dnazyme)是一种在金属离子协同存在情况下的具有催化功能的dna序列,具有特定序列和三维结构,能够进行特定的化学反应。其中了解最多的dna酶(dnazyme)是10
–
23,该酶是通过体外筛选的方式获得(santoro and joyce 1997),它能够切割靶标rna,该过程依赖于mg
2+
。10
–
23包含一个15个核苷酸的催化结构域,该结构域的两侧为底物结合臂,底物结合臂能使10
–
23精确识别靶标rna,底物结合臂的长度可根据rna底物的序列而变化。当10
–
23与底物结合后,在预定的嘌呤-嘧啶连接处进行rna切割,并且在g-u二核苷酸处观察到最高的剪切活性dnazyme。因其具有位点特异的切割rna的特性,可通过化学合成,易于改造和迭代更新,并且可针对不同的靶标rna进行重新设计。此外,dnazyme只需要mg
2+
就可获得较高酶活性,不需要其他辅助蛋白的作用,检测体系高效稳定。dnazyme通过结合臂与靶标rna的互补配对作用可精确识别靶标,具有较高的精准性。dnazyme未来或可用于生物工程、药物治疗、精准医疗等多个领域。
4.外泌体的研究发展依赖可靠稳定的提取技术和准确高效的检测技术。活体样本的外泌体不仅体积小,数量也极少,这使外泌体的分离和纯化非常困难。
5.目前最常用的外泌体分离方法主要有超速离心法、密度梯度超速离心法、peg-base沉淀法、磁珠免疫法。尽管这些方法在外泌体分离应用中都取得一定的进展,但同时也存在一些问题。例如,超速离心法是目前提取外泌体最常用的方法,也被认为是外泌体分离的“金标准”。根据大小差异,可采用超速离心法分离外泌体。该方法单次提取量高,成本低,但处理时间长,设备昂贵,回收率低,纯度低,可能导致非外泌体成分(如杂质蛋白和病毒)聚集。此外,反复离心可引起外泌体的损伤,导致外泌体的降解;密度梯度超速离心法能去除样品中的蛋白质污染,保持其活性和形态。然而,该方法工作量大,步骤复杂,耗时,回收率低;peg-base沉淀法是在一定盐浓度下向样品中加入亲水性聚乙二醇的分离方法。水分
子的结合降低了溶质的溶解度,以致在低速离心条件下外泌体出现凝结和沉淀。该方法被常被应用于沉淀外泌体,但也存在一些问题:比如外泌体的纯度和回收率低,机械力或化学添加物对外泌体产生破坏等;而磁珠免疫法则由于虽然免疫亲和技术具有高特异性、可获得高纯度的外泌体、且不影响外泌体形态完整等优点,是富集表征独特的外泌体的优选方法。但是此方法效率低,外泌体内含物的生物活性易受ph和盐浓度影响,不利于下游实验的进行。超速离心法在一定程度上能够规避上述方法存在的问题,因此应用更加广泛,是最常用的外泌体纯化手段。其该方法存在一些不足,主要包括:i)超速离心过程耗时耗力,且需要耗费大量的原料;ii)重复离心操作很有可能对外泌体的囊泡造成损害从而降低其质量;iii)操作条件苛刻,超高速离心机价格昂贵,并非所有实验室能够负担。因此如何简化外泌体的提取过程、减少对超高速离心机等大型昂贵仪器的依赖,如何进一步提高外泌体分离富集的效率,以及在高效富集的同时可以实现外泌体的准确定量成为亟需解决的问题。
技术实现要素:
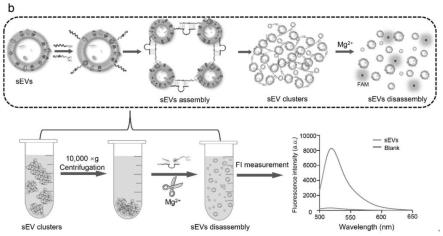
6.本发明提出了一种dnazyme触发的组装和拆卸系统,该系统可将单个纳米级sev相互偶联转化为簇,进而通过普通离心便能方便快捷地富集外泌体,然后在镁离子存在下分解成单个sev,通过记录核酸裂解时荧光的增加,实现sev的同时定量。
7.本发明设计了一种胆固醇尾端修饰的dnazyme探针和一种cd63适体缀合的底物探针,它们可以特异性地锚定在sev的膜表面。dnazyme探针和cd63适体缀合的底物探针的链之间的杂交触发了用于sev富集的三维dnazyme复合物的形成,在mg
2+
存在下可以将其切割以释放富集的sev。这些分离的sev可以通过在底物探针中使用荧光素-猝灭剂对进行标记来准确量化,从而在切割后实现可控的荧光释放。
8.本发明的目的之一在于提供一种用于分离外泌体的探针组,所述探针组包括2个探针,第1个探针的具体序列如seq id no.2、seq id no.3或seq id no.4所示,第2个探针的具体序列如seq id no.7所示。
9.本发明的目的之二在于提供一种外泌体分离富集方法,所述外泌体分离富集方法包括以下步骤:
10.(1)将权利要求1中的第1个探针和权利要求1中的第2个探针添加到sevs溶液中,将混合液置于室温下轻轻摇动;
11.(2)利用pbs缓冲液清洗数次sevs的团簇物质,并用pbs缓冲液重悬;
12.(3)再将mgcl2加入到重悬的sevs团簇样品中孵育;
13.(4)回收外泌体。
14.优选地,所述步骤(1)中第1个探针和第2个探针的终浓度都为100nm。
15.更优选地,所述步骤(1)中第1个探针和第2个探针的添加量都为5μl;sevs溶液的体积为1ml,浓度为1.0
×
107颗粒/微升。
16.更优选地,所述步骤(1)中室温下摇动时间为10分钟。
17.更优选地,所述步骤(3)中mgcl2的终浓度为40mm。
18.更优选地,所述步骤(3)中37℃孵育30分钟。
19.本发明的目的之三在于提供上述探针组在分离外泌体中应用。
20.本发明的目的之四在于提供上述外泌体分离富集方法在分离外泌体中应用。
21.与现有技术相比,本发明具有如下有益效果:
22.(1)相比于传统的超高速离心的外泌体分离方法,本发明利用成本更低,灵活高效的dnazyme核酸序列。将外泌体率先连接成簇,通过低速离心便可将外泌体分离富集下来,无需超高速离心,可实现免除大型仪器,方便高效的外泌体分离富集。
23.(2)在此基础上,再利用dnazyme空间结构的特异性切割,在镁离子存在下分解成单个sev。通过记录核酸裂解时荧光的增加,实现了sev的同时定量。整个分析过程可在1.5小时内完成,快捷高效。
24.(3)相对于传统试剂盒仅仅捕获外泌体的方法,我们对此进一步创新,解决了外泌体快速分离同时的检测问题。同时设计相关荧光探针在外泌体捕获序列上,在对外泌体进行捕获富集的同时,通过对荧光的测量,同时实现对外泌体量的检测。
25.(4)本发明实现了在无需超高速离心的条件下进行外泌体的分离富集;同时在捕获探针序列上设计应用荧光探针与淬灭基团,实现对外泌体的高度特异性富集检测的同时,通过特异性dnazyme的切割,达到外泌体富集分离同步检测的目的。
26.(5)建立了一种无需超高速离心、快速、高效外泌体的分离检测新技术,为外泌体的分离富集同步检测提供技术支撑。
附图说明
27.图1为本技术的试验原理图。
28.图2中a为实施例2探针序列结构及剪切位点示意图。
29.图2中b为实施例2page电泳图表征剪切图。
30.图2中c为实施例3荧光检测剪切反应情况图。
31.图2中d为实施例3反应的时间优化检测结果。
32.图2中e为实施例3反应的温度优化。
33.图3为实施例4中底物链substrate在sevs膜上锚定的实验原理与验证,其中(a)通过cd63核酸适配体识别底物substrate探针与sevs膜结合示意图。(b)底物链substrate探针锚定sevs的共聚焦显微镜分析。用pkh67染料将sevs膜染色(绿色),底物链标记cd63核酸适配体与cy5荧光基团(红色)。比例尺为10μm。
34.图4为实施例4中dnazyme探针在sevs膜上的锚定实验原理与验证,其中(a)链接了胆固醇分子的dnazyme探针识别锚定sevs膜的示意图。(b)dnazyme核酸链锚定sevs的共聚焦显微镜分析。用pkh67染料将sevs膜染色(绿色),dnazyme核酸链链接胆固醇分子并用cy5荧光基团(黄色)标记。比例尺为10μm。
35.图5中a为实施例5外泌体聚集成团及荧光标记。
36.图5中b为实施例5substrate探针锚定荧光cy5,共聚焦下荧光聚集结果图,pkh67是染色外泌体的标记物。
37.图5中c为实施例6囊泡富集后剪切分离原理示意图。
38.图5中d为实施例6不同浓度的外泌体检测荧光图。
39.图5中e为实施例6线性曲线分析。
40.图6为实施例7分离的sevs的形态与内容物的表征与分析结果,其中(a)本方法分离的sevs具有代表性的透射电镜tem图像。比例尺为100nm。(b)对照组rpmi-1640培养基(不
含fbs),实验组sevs和a549细胞(总蛋白)的几种代表性蛋白hsp70、cd63、cd9和canx蛋白进行western blotting分析。
41.图7为实施例7本发明对比超高速离心方法进行分离的sevs的nta分析,其中(a)通过本方法获得sevs的纳米颗粒跟踪分析(nta)。(b)常规超高速离心分离的sevs的nta分析。
具体实施方式
42.实施例1探针序列设计
43.本研究方法中所涉及的主要核酸探针序列结构如下表所示(表1),所有实验中反应体系的探针序列均由自主设计,本研究所有的实验所用核酸探针序列购自上海生工生物工程有限公司(中国)。
44.表1本发明所涉及的寡核苷酸探针序列
[0045][0046]
实施例2探针体系可行性验证
[0047]
本研究dnazymes体系的体外可行性验证实验的反应结果,首先配置该实验所用的核酸探针样品分为dnazyme探针(seq id no.5)、substrate(seq id no.2)、substrate(seq id no.2)+mgcl2、dnazyme(seq id no.5)+substrate(seq id no.2)、dnazyme(seq id no.5)+substrate(seq id no.2)+mgcl2(实验组),所有探针各自的浓度均为100nm,所有组的溶液均采用tm缓冲液,ph=7.2-7.4,mg
2+
终浓度为40mm。所有探针体系在仪器金属浴中进行自组装反应,反应条件为37℃,30分钟,参考以往的研究,mg
2+
均初步设置为40mm。利用不同核酸探针的dna分子量大小不同的特性,本结果利用聚丙烯酰胺凝胶电泳(page)验证了dnazyme与substrate核酸探针体系的反应组装的可行性,并验证其在镁离子作用下的裂解的可行性(通过释放荧光检测)。
[0048]
dnazymes反应体系的可行性实验需要用page电泳凝胶实验进行验证。在洗净的烧杯中配置12%聚丙烯酰胺凝胶,详细组成为:4ml丙烯酰胺、2ml 5
×
tbe缓冲液、4ml超纯水、20μltemed和150μl10%aps溶液(实验前配置,此溶液需要现配现用),使用1ml加样枪将以上混合样品进行充分混匀后,缓慢加入安装固定好的赶紧胶板模具中。加入完混合样品后按照需要立即插入洗净晾干的梳子模具。整个胶板于37℃放置30分钟使胶体充分凝固。最后将凝固好的胶板样本拆卸,随后组装好实验电泳槽装置,在电泳槽内缓慢加入干净的1
×
tbe缓冲液作为电泳缓冲液使用。在胶板的首个加样孔中加入指示条带dna ladder(其大小为50-500bp与20-500bp),将所有实验样品与对照样品使用dna loading buffer缓冲液混合均匀,混合比例为1:5。用加样枪将样本与dna loading buffer缓冲液进行充分混匀后,
再缓慢地加入到加样孔中。组装好电泳仪器设备,电泳实验先80v电压下,进行5分钟电泳实验。再通过在100v的电压,进行电泳40分钟实验,待dna ladder指示条带在整块胶板的三分之二处,关闭程序和仪器电源,停止电泳实验。拆卸装置取出凝胶,之后将凝胶在核酸染料下进行室温染色20分钟左右,可放在摇床上缓慢摇晃使染色均匀,该过程需要避光处理。最后使用凝胶成像仪观察实验结果。
[0049]
图2中a为探针序列结构及剪切位点示意图;b为page电泳图表征剪切。
[0050]
实施例3实验反应条件的优化
[0051]
本部分为dnazymes体系反应时间、镁离子浓度和温度的优化的体外表征实验。本实验的反应体系为:dnazyme(seq id no.5)+substrate(seq id no.3)+mgcl2+tm缓冲液(ph=7.2-7.4),时间实验优化部分的反应终止时间分别是:15分钟、20分钟、25分钟、30分钟、35分钟、40分钟和45分钟,镁离子浓度均为40mm,结果表明在反应在30分钟已达到有效的切割和信号放大。本实验部分所用的substrate核酸探针链中间部分用fam-bhq1进行标记。实验开始前首先检查调整好荧光光谱仪机器,且仪器需要提前15-30分钟打开,使激光发射器预热。然后将400μl待测样本液体加入到石英比色皿中,使用荧光光谱仪连续测量fi,激发波长为488nm,发射波长为520nm,电压为700v。同样方法,该反应体系在20-45℃的不同反应温度下进行温度优化实验,以确定该反应的最佳反应温度,结果显示最佳的探针反应浓度为37℃。同理在镁离子浓度10-60mm(10mm,20mm,30mm,40mm,50mm,60mm),验证该dnazymes反应体系荧光强度随镁离子的依赖性的变化趋势,验证该反应体系最优的镁离子反应浓度,本实验部分的反应体系中探针的最终浓度都为100nm,数据结果显示最佳的探针反应浓度为40mm。
[0052]
图2中c为荧光检测剪切反应情况;d为反应的时间优化检测;e为反应的温度优化。
[0053]
实施例4 dnazyme链和底物链substrate在sevs上的锚定实验
[0054]
在无菌环境下配制含有10%胎牛血清(fetal bovine serum,fbs)和1%青霉素链霉素的培养基,本实验使用a549非小细胞肺癌细胞作为囊泡提取的源细胞,细胞在含有配制好的培养基中,置于5%co2的湿润环境,恒温37℃培养。
[0055]
本实验中前期需要分离提取sevs进行可行性验证。该sevs由a549非小细胞肺癌细胞中分离提取。该部分实验首先待培养的a549非小细胞肺癌细胞生长到70%以上,用10ml无菌pbs缓冲液冲洗培养瓶中细胞三次,然后在不含胎牛血清的培养基中培养12小时以收集细胞上清液。在sevs的分离纯化过程中,首先收集上述a549非小细胞肺癌细胞在培养瓶中培养液,将收集的培养液在300
×
g离心10分钟,分离去除细胞颗粒。取上清液在1000
×
g离心10分钟,去除凋亡小体和碎片杂质。然后再次取上清液在10,000
×
g离心30分钟,去除大型细胞碎片等杂质。最后将所得上清液经0.22μm孔过滤器过滤收集。再次将过滤收集好的液体,于超高速离心机中,4℃温度下100,000
×
g离心1小时10分钟以沉淀底物。离心完成后弃掉上清。用无菌pbs缓冲液轻柔地清洗离心管底端几次,再次将洗涤收集的沉淀样品,4℃温度下100,000
×
g离心1小时十分钟以沉淀sevs。用4℃缓冲液pbs再次冲洗试管底部,以重新悬浮sevs。提取的sevs结合需要置于负80℃冰箱保存,备用于后续实验。
[0056]
利用激光共聚焦实验观察dnazyme和底物链substrate在sevs膜表面上的锚定效果。利用荧光染料pkh67将单独的sev进行染色,反应条件为室温下染色10分钟,染色完成后需要使用牛血清白蛋白终止sevs的染色反应,并且需要将染色后的sevs在100,000
×
g超速
4700进行测量统计。根据实验测量的荧光数值的变化情况,即可以定量检测出dnazyme核酸探针与底物链substrate核酸探针是否进行了裂解反应,以及本dnazymes体系的裂解活性。
[0064]
图5中c为囊泡富集后剪切分离原理示意图;d为荧光检测,不同浓度的外泌体检测荧光图;e为线性曲线分析。
[0065]
实施例7分离sevs的基本表征分析
[0066]
通过实验进一步对本策略分离后的sevs的形貌完整性与内容物完整性进行了实验验证。首先对提取的sevs的结构和蛋白进行了表征。通过透射电镜的结果表明提取的sevs膜结构是否完好。本研究进一步验证了该方法提取的sevs内容物的完整性,即蛋白分子的验证实验。该方法提取的sevs与a549细胞(总蛋白)的蛋白对照结果进行分析比较,选取rpmi-1640培养基(不含fbs)样品为对照组。本研究选用了hsp70、cd63蛋白、cd9蛋白作为对照实验。同时选取canx蛋白用以验证该策略提取的sevs的纯度问题。western blotting实验结果表明与a549细胞总蛋白的几种代表性蛋白相比,该方法提取的sevs保留了特异性蛋白,即hsp70蛋白、cd63蛋白、cd9蛋白和canx蛋白,且canx蛋白分析该技术提取的sevs纯度较好。本研究还探究了该方法提取的sevs数量对比传统的超高速离心机方法提取的sevs数量的nta粒径分析。
[0067]
图6中本研究策略分离的sevs的形态与内容物的表征与分析。(a)本方法分离的sevs具有代表性的透射电镜tem图像。比例尺为100nm。(b)对照组rpmi-1640培养基(不含fbs),实验组sevs和a549细胞(总蛋白)的几种代表性蛋白hsp70、cd63、cd9和canx蛋白进行westernblotting分析。
[0068]
图7中该策略对比超高速离心方法进行分离的sevs的nta分析。(a)通过本方法获得sevs的纳米颗粒跟踪分析(nta)。(b)常规超高速离心分离的sevs的nta分析。
[0069]
本研究中所有实验结果均以独立实验平均值
±
标准误差表示。所有实验数据采用配对的t检验(两组)或方差分析(三组或以上多组),以p<0.05作为显著性差异阈值。所有实验数据通过bonferroni进行校正。所有数据统计检验均使用graphpad prism 8.02软件进行计算。
[0070]
以上所述的实施例仅是对本发明的优选方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1