一种含GNA结构的起始加帽寡核苷酸引物及其制备方法和应用与流程
一种含gna结构的起始加帽寡核苷酸引物及其制备方法和应用
技术领域
1.本发明涉及化学及生物工程技术领域,具体涉及一种含gna结构的起始加帽寡核苷酸引物及其制备方法和应用。
背景技术:
2.在真核细胞中,大多数信使rna(mrna)的5'末端被封闭,或“帽化(加帽)”,所述帽包含有在两个核苷部分之间的5'-5'三磷酸键合和远端鸟嘌呤环上的7-甲基,mrna的帽化促进其在细胞中的正常功能。通过体外转录合成mrna已经成为引入外源基因并进行表达蛋白的重要工具,并广泛应用于疾病的治疗和预防中,体外转录合成mrna使得工作人员能够制备在各种生物学应用中表现适当的rna分子。此类应用包括多肽的研究应用和商业生产,例如,在无细胞翻译体系中产生在特定位点包含“非天然”氨基酸的多肽,或在培养的细胞中产生就其活性或稳定性而言需要翻译后修饰的多肽。在后者体系中,合成进行显著更长的时间,并因此产生更多的蛋白质。
3.专利cn201680067458.6报道了用于合成5
’‑
加帽rna的组合物和方法。其中起始加帽寡核苷酸引物具有通式形式m7gppp[n2’
ome
]n[n]m,其中m7g为n
7-甲基化的鸟苷或任何鸟苷类似物,n为任何天然的、修饰的或非天然的核苷,“n”可以是从0至4的任何整数且“m”可以是从1至9的整数。cleancap属于cap1,与arca 使用二聚体(m7gpppg)启动t7转录不同,cleancap使用三聚体(m7gpppamg)启动 t7转录。该方法的产量比较高,每毫升转录反应体系制备4mg加帽的rna,加帽效率可达90%,其转录产物的免疫原性低于arca。目前,mrna的免疫原性问题依然是该研究领域的重点。5’位帽子结构是降低免疫原性的关键结构,因此还需要设计更多的帽子结构来更好的降低免疫原性。
[0004]5’
位加帽类似物是降低mrna免疫原性的关键结构,通过优化帽类似物的结构可以降低细胞炎症因子表达水平。因此,需要不断优化帽类似物的结构,发现比目前已有的cleancap免疫原性更低、细胞炎症因子表达水平更低的新型帽类似物。
技术实现要素:
[0005]
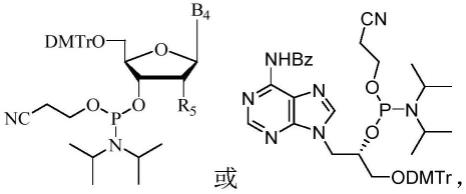
本技术提供了一种含gna结构的起始加帽寡核苷酸引物及其制备方法和应用,该含gna结构的起始加帽寡核苷酸引物结构中含有gna结构替换原有的五元糖环结构,替换后由于gna与炎症通路相关的蛋白结合能力弱,可以降低诱导的炎症因子表达。同时gna结构的帽类似物合成的mrna具有显著降低细胞炎症因子表达水平,更低的免疫原性。
[0006]
一种含gna结构的起始加帽寡核苷酸引物,其包含以下结构:
[0007][0008]
其中,x1、x2和x3分别独立的为o、ch2或nh;
[0009]
y1、y2和y3分别独立的为o、s、se或bh3;
[0010]
ra为rb为且当ra为时,
[0011]
rb为当ra为时,rb为
[0012]
r1、r2、r3独立的为h、oh、烷基、o-烷基、卤素;
[0013]
r4为氢、羟基、取代或未取代的o-烷基、取代或未取代的s-烷基、取代或未取代的nh-烷基、取代或未取代的n-二烃基、取代或未取代的o-芳香基、取代或未取代的s-芳香基、取代或未取代的nh-芳香基、取代或未取代的o-芳烷基、取代或未取代的s-芳烷基、取代或未取代的nh-芳烷基;
[0014]
b1、b2和b3独立的为天然的、或修饰的、或非天然的核苷碱基。
[0015]
含gna结构的起始加帽寡核苷酸引物的制备方法,包括以下步骤:(1) 二磷酸咪唑中间体(化合物21)的合成:以n2-异丁酰鸟嘌呤(化合物14)为起始原料,依次进行糖苷化、二磷酸化、n7的甲基化、多磷酸的咪唑化等反应,合成二磷酸咪唑中间体;(2)磷酸酯键连接的二核苷酸的制备:通过亚磷酰胺单体与双取代核苷单体,在四氮唑的作用下偶联形成第一个磷酸酯键,通过酸作用,脱除保护基,然后引入第二个磷酸,最终水解得到磷酸酯键连接的二核苷酸;(3)含gna结构的起始加帽寡核苷酸引物的合成:二磷酸咪唑中间体与磷酸酯键连接的二核苷酸反应制备含gna结构的起始加帽寡核苷酸引物;
[0016]
上述亚磷酰胺单体结构式为:
[0017]
其中,r5为h、oh、烷基、o-烷基、卤素;b4为天然的、或修饰的、或非天然的核苷碱基。
[0018]
上述双取代核苷单体选自中的任一种。
[0019]
该含gna结构的起始加帽寡核苷酸引物的制备方法,具体包括以下步骤:
[0020]
(1)化合物21的合成:
[0021]
(1-1)称取n2-异丁酰鸟嘌呤(化合物14)溶解在乙腈中,搅拌均匀澄清后加入双乙酰化保护的甘油类似物,充分混合后,加入bsa和四氯化锡,室温反应 5h后hplc监控反应,反应结束后,加入饱和氯化铵溶液猝灭反应,并用乙酸乙酯萃取反应体系三次,乙酸乙酯相干燥旋干后柱分离得产物15;
[0022]
(1-2)称取化合物15,溶解在甲醇中,加入氨水,反应结束后旋干体系,并用 2体积的乙腈带水,水分小于100ppm后,再加入乙腈,吡啶,冰浴至反应体系温度小于4℃,缓慢滴加三甲基氯硅烷,滴加结束后加入苯甲酰氯,室温反应5h 后,浓缩的目标化合物16;
[0023]
(1-3)取化合物16溶解在吡啶中,冰浴下分批将tscl加入到反应液中,随后升温至室温反应。tlc监测反应至完全。将反应液浓缩,粗品用柱层析纯化得到化合物17;
[0024]
(1-4)取化合物17溶解在dmf中,将三(四丁基)焦磷酸铵加入到反应液中,随后室温反应20小时。hplc监控反应完全。减压浓缩除去大部分溶剂后得到化合物18粗品。将化合物18粗品溶解在浓氨水继续室温搅拌2小时。hplc监控反应完全。浓缩除去氨水后用去离子水稀释。将混合物用deae sephadex,流动相用0-1.0m的teab洗脱液线性梯度洗脱得到化合物19;
[0025]
(1-5)称取化合物19溶解在水溶液,反应液冷却至4℃,缓慢滴加硫酸二甲酯,过程中用2m的氢氧化钠调节ph不超过5,hplc监测反应,反应结束后离子色谱纯化得化合物20;
[0026]
(1-6)将化合物20溶解在dmf中,与三苯基膦,二硫二吡啶,咪唑充分反应,反应液加入4m的高氯酸钠丙酮溶液中析出,滤饼用丙酮充分洗涤得目标化合物 21。
[0027]
(2)磷酸酯键连接的二核苷酸的制备:
[0028]
称取亚磷酰胺单体于单口瓶中,用二氯甲烷溶解,再加入双取代核苷单体,降温至25
±
2℃,氮气鼓吹下加入四氮唑,升温至25
±
2℃反应。监测反应结束后,将碘吡啶溶液加入到反应液中,监测反应结束后旋干,浓缩后的油膏溶解在二氯甲烷中,加入三氟乙酸,监测反应结束后,旋干,石油醚/二氯甲烷按一定比例打浆,过滤得中间体;将中间体溶解在磷酸三甲酯中,加入三氯氧磷充分搅拌反应,监测反应结束后,在旋瓶中加入甲醇和浓氨水,
室温反应4小时,监测反应,反应结束后旋干,加入超纯水,进入反向离子渗透设备,洗涤浓缩,冻干得目标化合物;
[0029]
(3)含gna结构的起始加帽寡核苷酸引物的合成:
[0030]
将化合物21溶解在含有mncl2(0.2mol)的dmf溶液中,并添加到磷酸酯键连接的二核苷酸的dmf溶液中。在室温下搅拌反应。24小时后,用0.25m edta 溶液终止反应。将混合物装载到deaesephadex柱(30
×
500cm)上。将产物使用 0-1.0m的teab洗脱液线性梯度洗脱。收集hplc纯度>97%的洗脱产物,浓缩完以上分离液,再装载到强阴离子树脂,使用0-1.0m的醋酸钠洗脱液线性梯度洗脱,收集hplc纯度>98.5%的洗脱产物,合并高纯度洗脱液,通过纳滤设备去除残留的醋酸钠溶液并浓缩得目标产物含gna结构的起始加帽寡核苷酸引物。
[0031]
该含gna结构的起始加帽寡核苷酸引物用于t7 rna聚合酶体系下的 mrna加帽。t7 rna聚合酶是一种依赖dna的rna聚合酶,其对噬菌体 t7启动子序列有高特异性。该酶从t7启动子插入到转录载体下游的dna上合成大量rna。t7 rna聚合酶催化的ivt(体外转录)反应体系是目前最成熟的mrna制备体系。
[0032]
通常20ul的ivt反应体系中含有50u的t7 rna聚合酶,同时搭配1ul的帽类似物(100mm)可以获得最佳的转录产量以及加帽效率。
[0033]
本发明提供了一种含gna结构的起始加帽寡核苷酸引物及其制备方法和应用,该含gna结构的起始加帽寡核苷酸引物适用于以dna序列为模板利用体外共转录方法生产的mrna,该dna序列可以来源或改造自病毒、动物、植物等物种,同时其生产的mrna具有更低的免疫原性、更高的蛋白翻译效率、更好的稳定性。
[0034]
本发明相比现有技术具有以下优点:
[0035]
与现有帽结构类似物cleancap相比,本发明的含gna结构的起始加帽寡核苷酸引物其生产的mrna具有显著降低细胞炎症因子表达水平,更低的免疫原性。
具体实施方式
[0036]
下面将结合本发明实施例,对本发明的技术方案进行清楚、完整的描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0037]
各实施例中所使用的原料名称及来源参见下表1:
[0038]
表1
[0039]
[0040]
[0041][0042]
实施例1:以化合物9和化合物n为原料的含gna结构的起始加帽寡核苷酸引物的合成方法
[0043]
将化合物n溶解在含有mncl2(0.2mol)的dmf溶液中,并添加到化合物 9(1.8mol)的dmf溶液中。在室温下搅拌反应。24小时后,用10l的0.25m edta 溶液终止反应。将混合物装载到deaesephadex柱(30
×
500cm)上。将产物使用 0-1.0m的teab洗脱液线性梯度洗脱。收集hplc纯度>97%的洗脱产物,浓缩完以上分离液,再装载到强阴离子树脂,使用0-1.0m的醋酸钠洗脱液线性梯度洗脱,收集hplc纯度>98.5%的洗脱产物,合并高纯度洗脱液,通过纳滤设备去除残留的醋酸钠溶液并浓缩得目标产物gna-1,反应路线流程,如下方程式(4):
[0044][0045]
其中,化合物9通过以下步骤制备得到:
[0046]
(1-1)称取50g化合物1溶解在200ml乙腈中,搅拌均匀澄清后加入1.2eq的双乙酰化保护的甘油类似物,充分混合后,加入1.2eq bsa和1.4eq的四氯化锡,室温反应5h后hplc监控反应,反应结束后,加入300ml饱和氯化铵溶液猝灭反应,并用乙酸乙酯萃取反应体系三次,乙酸乙酯相干燥旋干后柱分离得产物2;
[0047]
(1-2)称取10g化合物2,溶解在20ml甲醇中,加入2eq的氨水,反应结束后旋干体系,并用2体积的乙腈带水,水分小于100ppm后,再加入2体积的乙腈,2eq的吡啶,冰浴至反应体系温度小于4℃,缓慢滴加2eq三甲基氯硅烷,滴加结束后加入1.2eq的苯甲酰氯,室温反应5h后,浓缩的目标化合物3,纯化后通过与四氮唑和膦试剂反应得到目标化合物4;
[0048]
(1-3)称取50g 4亚磷酰胺单体于单口瓶中,用200ml的二氯甲烷溶解,再加入1.2eq 2’,3’乙酰基鸟苷,降温至25
±
2℃,氮气鼓吹下加入0.5eq四氮唑,升温至25
±
2℃反应。监测反应结束后,将1.2eq的碘吡啶溶液加入到反应液中,监测反应结束后旋干,浓缩后的油膏溶解在400ml二氯甲烷中,加入1.1eq的三氟乙酸,监测反应结束后,旋干,石油醚/二氯甲烷按一定比例打浆,过滤得化合物7;将化合物7溶解在400ml磷酸三甲酯中,加入1.2eq
的三氯氧磷充分搅拌反应,监测反应结束后,在旋瓶中加入300ml甲醇和300ml浓氨水,室温反应4小时,监测反应,反应结束后旋干,加入20l超纯水,进入反向离子渗透设备,洗涤浓缩,冻干得目标化合物9;
[0049]
化合物9具体的反应路线流程,如下方程式(1):
[0050][0051]
其中,化合物n通过以下步骤得到:1)称取5g鸟苷,分散在50ml的dmf 中,冰浴使反应液内温低于10℃,分两个批次加入1.2eq的tbscl,hplc监控反应至原料≤5%,反应结束后加入100ml的水将产物析出,过滤并洗涤滤饼;将2g滤饼溶解在10ml的磷酸三甲酯中,反应液冷却至0℃,缓慢滴加1.2eq三氯氧磷,低温反应4小时后,加入2m的醋酸铵溶液猝灭反应,反相色谱纯化得目标化合物f,得到的化合物f与1eq的三苯基膦,2eq的二硫二吡啶,4eq的咪唑充分反应,反应液加入4m的高氯酸钠丙酮溶液中析出,滤饼用丙酮充分洗涤得目标化合物g;
[0052]
2)称取2g目标化合物g溶解dmf中,加入3eq的磷酸三丁胺,充分搅拌得目标化合物h,向反应液中加入20eq的水溶液,反应液冷却至4℃,缓慢滴加硫酸二甲酯,过程中用2m的氢氧化钠调节ph不超过5,hplc监测反应,反应结束后离子色谱纯化得目标化合物i;
[0053]
3)将4g化合物i溶解在50ml dmf中,与1eq的三苯基膦,2eq的二硫二吡啶, 4eq的咪唑充分反应,反应液加入4m的高氯酸钠丙酮溶液中析出,滤饼用丙酮充分洗涤得目标化合物n。
[0054]
化合物n反应路线流程,如下方程式(7):
[0055][0056]
实施例2:以化合物21和a-g-p为原料的含gna结构的起始加帽寡核苷酸引物的合
成方法
[0057]
以化合物21和a-g-p为原料,参考实施例1目标产物gna-1的合成方法得到实施例2的起始加帽寡核苷酸引物gna-2。反应路线流程,如下方程式(5):
[0058][0059]
其中,化合物21的制备包括以下步骤:1)化合物15的合成参考化合物2的合成步骤;2)化合物16的合成参考化合物3的合成步骤;3)取10g化合物16 溶解在吡啶(100ml)中,冰浴下分批将tscl(1.2eq.)加入到反应液中,随后升温至室温反应。tlc监测反应至完全。将反应液浓缩,粗品用柱层析纯化得到化合物17;4)取5g化合物17溶解在dmf(50ml)中,将三(四丁基)焦磷酸铵(2.0eq.)加入到反应液中,随后室温反应20小时。hplc监控反应完全。减压浓缩除去大部分溶剂后得到化合物18粗品。将化合物18粗品溶解在150ml 浓氨水继续室温搅拌2小时。hplc监控反应完全。浓缩除去氨水后用去离子水稀释。将混合物用deae sephadex,流动相用0-1.0m的teab洗脱液线性梯度洗脱得到化合物19;5)化合物20的合成参考化合物11的合成步骤;6)化合物21的合成参考化合物12的合成步骤。
[0060]
化合物21的反应路线流程,如下方程式(3):
[0061][0062]
其中,a-g-p的合成路线为:称取5kg的2’ome-ra亚磷酰胺单体于单口瓶中,用50l的二氯甲烷溶解,再加入2.73kg的2’,3’乙酰基鸟苷,降温至25
±
2℃,氮气鼓吹下加入880g四氮唑,升温至25
±
2℃反应。监测反应结束后,将1.2eq 的碘吡啶溶液加入到反应液中,监测反应结束后旋干,浓缩后的油膏溶解在4l 二氯甲烷中,加入1.1eq的三氟乙酸,监测反应结束后,旋干,石油醚/二氯甲烷按一定比例打浆,过滤得化合物g1;将g1溶解在4l乙腈中,加入1.2eq的膦试剂、1.2eq的四氮唑充分搅拌反应,监测反应结束后,再加入1.2eq的碘吡啶溶液加入到反应液中,监测反应结束后旋干,在旋瓶中加入3l甲醇和3l浓氨水,室温反应
4小时,监测反应,反应结束后旋干,加入20l超纯水,进入反向离子渗透设备,洗涤浓缩,冻干得目标化合物a-g-p,反应路线流程,如下方程式(2):
[0063][0064]
实施例3:以化合物9和化合物21为原料的含gna结构的起始加帽寡核苷酸引物的合成方法
[0065]
以化合物9和化合物21为原料(化合物9参考实施例1中的制备方法得到,化合物21参考实施例2的制备方法得到),参考实施例1目标产物gna-1的合成方法得到实施例3的起始加帽寡核苷酸引物gna-3。反应路线流程,如下方程式(5):
[0066][0067]
对比例1:
m7
gpppa2’
ome
pg
[0068]
m7
gpppa2’
ome
pg的合成方法参考上述实施例的合成方法(所使用的原料参考各实施例中的制备方法),反应路线流程,如下方程式(8):
[0069][0070]
各实施例所得到的含gna结构的起始加帽寡核苷酸引物以及对比例所得到的加帽类似物结构如下表2所示,
[0071]
表2
[0072][0073]
测试例1:mrna体外转录产量及加帽效率的测定
[0074]
利用含gna结构的起始加帽寡核苷酸引物进行mrna的体外合成:先用 noti线性化质粒,4℃酶切过夜;dna模板抽提;体外转录合成mrna,分别使用实施例1-3及对比例1的含gna结构的起始加帽寡核苷酸引物作为帽结构。
[0075]
反应体系如表3:
[0076]
表3
[0077]
体系用量t7 rna聚合酶50u10x buffer2μl
100mm atp1μl100mm gtp1μl100mm ctp1μl100mm n1-me-putp1μl100mm帽类似物1μl无机焦磷酸酶0.05u核酸酶抑制剂20u无菌无酶水补足至20μl模板1μg
[0078]
备注:在实验过程中,首先计算好体系所需物料体积,然后进行加样。首先在体系中加入无菌无酶水,随后依次加入10xbuffer、ntps、帽类似物,混匀后轻轻离心,随后加入核酸酶抑制剂、无机焦磷酸酶、t7rna聚合酶、线性化dna模板,充分混匀后轻轻离心,于37℃下孵育。2小时后加入dnasei1u,37℃继续孵育30分钟以去除dna模板,然后进行rna纯化,通常使用磁珠纯化方法。纯化的mrna用无菌无酶水进行溶解,随后利用nanodropone进行定量检测。
[0079]
液相色谱质谱法(lc-ms)被用来检测不同起始帽类似物的mrna的ivt加帽率;首先需要设计一段与转录产物mrna起始碱基匹配的具有标记的dna探针,通常的标记为biotin标记,将链霉亲和素标记的磁珠清洗后与合成的dna探针、mrna及10
×
rnasehreactionbuffer室温室温孵育30分钟,边孵育边缓慢混匀,随后加入20ulrnaseh(5u/ul)孵育37度3h,每半个小时混匀一次。孵育结束后对磁珠进行清洗,清洗完成后的磁珠加入100μl加热到80℃的75%甲醇,混合物在加热板上加热到80℃,保持3分钟,然后放置磁力架上吸取上清,使用蒸发离心机在室温下干燥45分钟至10μl。然后将样品重新悬浮在50μl的100μmedta/1%meoh中,即可用于lc-ms分析,确定转录反应中rna的加帽情况。由于加帽与非加帽的碱基在分子量上有明显区别,利用分子质量差别即可判定不同帽类似物起始的mrna转录的加帽率。具体结果见表4。
[0080]
表4
[0081]
编号产量(μg)加帽率(%)实施例15593.2实施例25091.3实施例34885.4对比例15692.0
[0082]
由实验结果可知,本技术的实施例1和实施例2含gna结构的起始加帽寡核苷酸引物与对比例相比具有相同水平的mrna体外转录产量以及加帽效率;但是实施例3的加帽效率有明显的降低,这可能它由于结构和帽类似物差距较大,不利于t7rna聚合酶的催化反应,导致加帽效率降低。
[0083]
测试例2:mrna在细胞内刺激炎症因子的表达
[0084]
hela细胞以4
×
105/孔的密度铺于6孔板,细胞密度大约为80%时进行转染。每孔转染2ugmrna,转染试剂为lipofectaminemessengermaxtransfectionreagent(invitrogen),转染步骤参照说明书进行。24小时后收集细胞,利用trizol抽取rna,并将
rna逆转录为cdna。最后利用实时定量荧光pcr检测细胞内炎症因子的表达,内参基因为β-actin。每个基因的检测需要重复三次,每个基因的表达结果为相对于对比例1帽类似物结果的相对值。数据表示为平均值
±
标准差,所得结果见下表5:
[0085]
表5
[0086]
编号il-6的表达infb的表达infa的表达mx1的表达实施例10.50.60.60.7实施例20.80.50.60.7实施例31.11.31.21.0对比例11.01.01.01.0
[0087]
由上表4的实验数据可知,本技术的实施例1和实施例2含gna结构的起始加帽寡核苷酸引物与对比例1相比具有显著降低细胞炎症因子表达水平,具有更低的免疫原性。但是实施例3的细胞炎症因子表达水平高于对比例1,这可能是由于加帽类似物加帽效率较低,产物中残留了未加帽的mrna,导致免疫原性增强。
[0088]
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1