一种高强度自修复弹性体材料及其制备方法
1.本发明涉及自修复材料领域,具体涉及一种高强度自修复弹性体材料及其制备方法。
背景技术:
2.自修复弹性体是通过可逆的动态非共价或/和动态共价键替代传统的不可逆的共价键,引入到高分子骨架中形成交联结构而制备得到的一类智能材料。自修复弹性体具有高弹性、高韧性、使用寿命长等突出优点,因此在柔性电子、昂贵涂层、生物医学材料、储核材料和航空航天等领域展现了广泛的应用前景。但是,动态化学键由于其本质的高活性和可逆性,键能往往较低,制备的材料强度偏弱。强度和自修复性能这对矛盾是该领域亟需解决的问题。
3.近年来,研究人员通过设计不同的动态结构来调节自修复弹性体材料的力学强度和修复性能。主要可以分为由动态非共价键,如氢键、配位、ππ堆叠等超分子相互作用交联的弹性体,如申请公布号为cn 109280143 a的专利设计了一种由金属配位键交联的自修复弹性体,以及由动态共价键,如酯键、硼酯键、氨基甲酸酯、烷氧基胺等交联的弹性体,如journal ofmaterials chemistry a(2019,7,1459-1467)报导了一种由甲硅烷基醚构成的共价交联弹性体。动态共价键的作用强度通常强于超分子相互作用,因此动态共价网络交联的自修复弹性体一般具有更高的力学性能和更可靠的使用性能。但通过动态共价键制备的自修复弹性体拉伸强度多小于10mpa,依旧处于一个相对较低的水平。
4.增大交联密度可进一步提高自修复弹性体的拉伸强度,如nano energy(2021,87,105822)报导的高交联弹性体,大量交联剂的添加,会损害弹性体固有的优异性质,如高弹性、低滞后性等等,更重要的是,高的交联密度会显著抑制分子链的活动能力,松弛时间大幅延长,导致弹性体的修复性能明显下降。因此,如何兼顾强度与自修复性能是该领域的首要技术难点。
技术实现要素:
5.为解决现有技术中的不足,本发明通过结合型动态键与弹性体反应构建动态共价交联网络,利用结合型动态键在活化温度以上可以实现键与键交换的特性;并且在活化温度以上,将弹性体进行拉伸,并固定应变,通过结合型动态键的交换实现应力松弛从而使得取向结构得到保持;取向后的弹性体在取向方向上拉伸强度显著提升,并且可以在相对温和的条件下实现良好的修复效果;即本发明得到了一种由结合型动态键交联的、具有取向结构的自修复弹性体;即得到了一种兼具高强度和自修复性能的弹性体材料。
6.本发明的技术方案:
7.本发明要解决的第一个技术问题是提供一种高强度自修复弹性体材料,所述弹性体材料的原料包括以下重量份的组分:环氧化弹性体或含不饱和双键的弹性体100份,结合型动态交联剂1~10份,催化剂0.1~1份;其中,所述结合型动态交联剂的结构式如式i所
示:
8.hs-r-sh
9.式i
10.式i中r选自苯硼酸酯或酯键。本发明中,所述结合型动态交联剂为可以与弹性体进行反应形成结合型动态键交联网络的物质。
11.进一步,所述的结合型动态交联剂选自:2,2
’‑
(1,4-亚苯基)-双[4-硫醇1,3,2-二氧硼杂环戊烷](bdb)、双(3-巯基丙酸)乙二酯、双(巯基乙酸)乙二醇酯、1,4-丁二醇双(巯基乙酸酯)或四(3-巯基丙酸)季戊四醇酯中的至少一种。
[0012]
进一步,所述环氧化弹性体选自:环氧化天然橡胶、环氧化乙丙橡胶、环氧化丁苯橡胶、环氧化丁基橡胶或环氧化顺丁橡胶中的至少一种。
[0013]
进一步,所述含不饱和双键的弹性体选自天然橡胶、丁苯橡胶、丁基橡胶、丁腈橡胶、异戊橡胶或顺丁橡胶中的至少一种。
[0014]
进一步,所述催化剂选自:4-二甲氨基吡啶、醋酸锌或钛酸四丁酯中的至少一种。
[0015]
本发明要解决的第二个技术问题是提供上述高强度自修复弹性体材料的制备方法,所述方法为:先将环氧化弹性体或含不饱和双键的弹性体、结合型动态交联剂和催化剂共混均匀得共混物;然后将共混物热压固化得固化物,固化过程中所述结合型动态交联剂通过硫醇与弹性体的双键或环氧基团进行点击反应形成动态键交联网络;最后将所得固化物在动态键的键交换反应温度以上拉伸至应变在100~1000%,使得弹性体的分子链发生取向,然后保持30~300min,制得了高强度的自修复弹性体材料。
[0016]
进一步,所述动态键的键交换反应温度为80℃~200℃。
[0017]
进一步,所述高强度自修复弹性体材料的制备方法为:先将环氧化弹性体或含不饱和双键的弹性体、结合型动态交联剂和催化剂共混均匀得共混物;然后将共混物热压固化得固化物,固化过程中所述结合型动态交联剂通过硫醇与弹性体的双键或环氧基团进行点击反应形成结合型动态键交联网络;最后将所得固化物在结合型动态键的键交换反应温度以上拉伸至应变在100~1000%,使得弹性体的分子链发生取向,然后保持30~300min,通过结合型动态键的键交换实现了弹性体内部应力的松弛,降低了由弹性体的回弹性所引起的取向回复,并结合交联网络的固定作用,最终实现了取向结构的固定,制得了高强度的自修复弹性体材料。
[0018]
进一步,上述方法中,各原料共混可以通过熔融共混或开炼共混。
[0019]
进一步,所述制备方法包括如下步骤:
[0020]
1)将环氧化弹性体或含不饱和双键的弹性体、结合型动态交联剂和催化剂通过熔融共混或开炼共混得共混物;各原料的比例为:环氧化弹性体或含不饱和双键的弹性体100重量份,结合型动态交联剂1~10重量份,催化剂0.1~1重量份;
[0021]
2)将步骤1)所得共混物于100~200℃、5~20mp的压力下进行热压固化30~180min得固化物;
[0022]
3)将步骤2)中所得固化物在80~200℃下拉伸至应变在100~1000%,使得弹性体分子链取向,保持30~300min得到所述高强度自修复弹性体材料。
[0023]
步骤1)中,所述的熔融共混设备为双辊开炼机或密炼机。
[0024]
步骤1)中,所述共混温度20~100℃,共混时间为5~15min。
[0025]
本发明要解决的第三个技术问题是提供一种提高自修复弹性体材料强度的方法,所述方法为:选择环氧化弹性体或含不饱和双键的弹性体为基体,在其中引入结合型动态交联剂和催化剂;先将各原料共混均匀得共混物,然后将共混物热压固化得固化物,最后将固化物通过拉伸处理即可;其中,各原料的比例为:环氧化弹性体或含不饱和双键的弹性体100重量份,结合型动态交联剂1~10重量份,催化剂0.1~1重量份;所述结合型动态交联剂的结构式如式i所示:
[0026]
hs-r-sh
[0027]
式i
[0028]
式i中r选自苯硼酸酯或酯键。
[0029]
进一步,所述热压固化的方法为:将所得共混物于100~200℃、5~20mp的压力下进行热压固化30~180min。
[0030]
进一步,所述将固化物拉伸处理的方法为:将所得固化物在80~200℃下拉伸至应变在100~1000%,使得弹性体分子链取向,保持30~300min。
[0031]
本发明的有益效果:
[0032]
(1)本发明利用活化温度以上结合型动态键的键交换特性,松弛掉弹性体内部存在的应力,从而固定弹性体分子链沿拉伸方向的取向,使得拉伸强度得到大幅度提升(表1)。
[0033]
(2)本发明采用结合型动态键交联的自修复弹性体,与传统的永久交联键相比,材料受损后,可以在温度场条件下实现修复;与超分子作用力相比,具有更高的力学强度和更可靠的使用性能(图2)。
[0034]
(3)通过对拉伸取向过程中的松弛行为进行控制可以实现弹性体强度在较宽范围内的调节(图3)。
附图说明:
[0035]
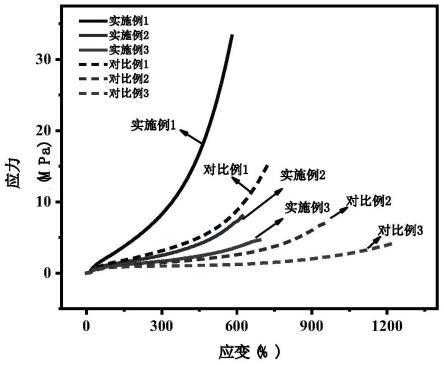
图1为本发明实施例1~3及对比例1~3的拉伸性能测试图,由图可知,随交联剂含量提高,弹性体强度不断升高,取向前后样品的强度差异增加;实施例1的拉伸强度超过30mpa,说明本发明所得弹性体具有优异的力学性能。
[0036]
图2为本发明实施例1~3及对比例1~3在80℃修复24h后的拉伸性能测试图,由图可知,修复后,拉伸取向后的弹性体较未拉伸取向的弹性体具有更高的力学强度,说明该弹性体具有良好的修复性能。
[0037]
图3为本发明实施例1、4、5的拉伸性能测试图,表明说明可以通过调控取向时间来制备具有高强度的自修复弹性体。
[0038]
图4为本发明的机理说明图,即弹性体的固化和取向过程示意图,由图可知,交联剂两端的巯基与弹性体的环氧官能团或不饱和碳碳双键发生反应,构筑交联网络;在由结合型动态共价键构筑的交联网络的键交换反应温度以上将弹性体拉伸至固定应变使得弹性体的分子链发生取向,然后保持适当时间;拉伸使得弹性体的分子链发生取向,通过所用结合型动态键的键交换实现弹性体内部应力的松弛,降低了弹性体的回弹性引起的取向回复,并结合交联网络的固定作用,最终实现实现了取向结构的固定,制得了高强度的自修复弹性体材料。
具体实施方式
[0039]
本发明提供一种高强度的自修复弹性体材料,由以下材料组成:含不饱和双键的弹性体或环氧化弹性体100份,结合型动态交联剂1-10份,催化剂0.1-1份;制备过程可采用下述方式:(1)将弹性体、结合型动态交联剂和催化剂熔融共混或开炼均匀;(2)将步骤(1)中混合物放于平板硫化仪中热压固化;(3)将步骤(2)中固化后产物在高温条件下拉伸至固定应变使得弹性体分子链取向,保持适当时间,通过结合型动态键松弛内应力将取向结构固定。本发明的自修复弹性体,由动态键交联固定分子链取向结构,可以显著提高弹性体的强度,同时兼具良好的自修复性能;即制得了一种结合型动态键交联的、具有取向结构的弹性体材料。
[0040]
本发明中,所述高强度自修复弹性体材料的制备过程中,所述结合型动态交联剂通过硫醇与弹性体的双键或环氧基团进行点击反应形成结合型动态键交联网络,由于所得结合型动态键在交换反应温度以上发生结合型交换反应,旧键断裂和新键形成同时发生,使得材料交联密度保持不变;以环氧化天然橡胶和bdb交联剂为例,交联反应及结合型交换反应式如下所示,交联剂bdb的巯基在dmap催化条件下与环氧天然橡胶上的环氧官能团发生反应,得到交联弹性体;在动态键的键交换温度以上硼酯键被激活,发生键间交换。
[0041][0042]
下面结合实施例对本发明的具体实施方式做进一步的描述,并不因此将本发明限制在所述的实例范围之中。
[0043]
实施例1
[0044]
将6.0g的1,4-苯二硼酸和8.02g的1-硫代甘油溶于200ml四氢呋喃中,加入10.0g无水硫酸镁,在25℃条件下搅拌24小时,过滤,除去无水硫酸镁,旋蒸,得到2,2
’‑
(1,4-亚苯基)-双[4-硫醇1,3,2-二氧硼杂环戊烷](bdb)。
[0045]
将环氧度30%的环氧化天然橡胶(环氧化异戊二烯)45g、bdb 4.5g、2-二甲氨基吡啶dmap 0.45g于双辊开炼机上在25℃条件下混合均匀;将混合物剪碎成小颗粒后放入平板
流变仪中热压固化,固化温度160℃,固化压力10mpa,固化时间45min。将固化后的样品裁成长60mm宽27mm的矩形样条,由拉伸夹具夹持,120℃条件下,将样条以10mm/min的拉伸速率拉至300%应变,固定应变,保持90min制得本发明高强度自修复弹性体。所得弹性体材料的拉伸强度及修复性能见表1,本发明实施例中,修复条件为在80℃修复24h。
[0046]
实施例2
[0047]
将6.0g 1,4-苯二硼酸和8.02g 1-硫代甘油溶于200ml四氢呋喃中,加入10.0g无水硫酸镁,在25℃条件下搅拌24小时,过滤,除去无水硫酸镁,旋蒸,得到2,2
’‑
(1,4-亚苯基)-双[4-硫醇1,3,2-二氧硼杂环戊烷](bdb)。
[0048]
将环氧度30%的环氧天然橡胶45g、bdb 2.25g、2-二甲氨基吡啶dmap 0.225g于双辊开炼机上在25℃条件下混合均匀。将混合物剪碎成小颗粒后放入平板流变仪中热压固化,固化温度160℃,固化压力10mpa,固化时间45min。将固化后的样品裁成长60mm宽27mm的矩形样条,由拉伸夹具夹持,120℃条件下,将样条以10mm/min的拉伸速率拉至300%应变,固定应变,保持90min。胶料性能及修复性能见表1。
[0049]
实施例3
[0050]
将6.0g 1,4-苯二硼酸和8.02g 1-硫代甘油溶于200ml四氢呋喃中,加入10.0g无水硫酸镁,在25℃条件下搅拌24小时,过滤,除去无水硫酸镁,旋蒸,得到2,2
’‑
(1,4-亚苯基)-双[4-硫醇1,3,2-二氧硼杂环戊烷](bdb)。
[0051]
将环氧度30%的环氧天然橡胶45g、bdb 0.45g、2-二甲氨基吡啶dmap 0.045g于双辊开炼机上在25℃条件下混合均匀。将混合物剪碎成小颗粒后放入平板流变仪中热压固化,固化温度160℃,固化压力10mpa,固化时间45min。将固化后的样品裁成长60mm宽27mm的矩形样条,由拉伸夹具夹持,120℃条件下,将样条以10mm/min的拉伸速率拉至300%应变,固定应变,保持90min。胶料性能及修复性能见表1。
[0052]
实施例4
[0053]
将6.0g 1,4-苯二硼酸和8.02g 1-硫代甘油溶于200ml四氢呋喃中,加入10.0g无水硫酸镁,在25℃条件下搅拌24小时,过滤,除去无水硫酸镁,旋蒸,得到2,2
’‑
(1,4-亚苯基)-双[4-硫醇1,3,2-二氧硼杂环戊烷](bdb)。
[0054]
将环氧度30%的环氧天然橡胶45g、bdb 4.5g、2-二甲氨基吡啶dmap 0.45g于双辊开炼机上在25℃条件下混合均匀。将混合物剪碎成小颗粒后放入平板流变仪中热压固化,固化温度160℃,固化压力10mpa,固化时间45min。将固化后的样品裁成长60mm宽27mm的矩形样条,由拉伸夹具夹持,120℃条件下,将样条以10mm/min的拉伸速率拉至300%应变,固定应变,保持30min。胶料性能见表1。
[0055]
实施例5
[0056]
将6.0g 1,4-苯二硼酸和8.02g 1-硫代甘油溶于200ml四氢呋喃中,加入10.0g无水硫酸镁,在25℃条件下搅拌24小时,过滤,除去无水硫酸镁,旋蒸,得到2,2
’‑
(1,4-亚苯基)-双[4-硫醇1,3,2-二氧硼杂环戊烷](bdb)。
[0057]
将环氧度30%的环氧天然橡胶45g、bdb 4.5g、2-二甲氨基吡啶dmap 0.45g于双辊开炼机上在25℃条件下混合均匀。将混合物剪碎成小颗粒后放入平板流变仪中热压固化,固化温度160℃,固化压力10mpa,固化时间45min。将固化后的样品裁成长60mm宽27mm的矩形样条,由拉伸夹具夹持,120℃条件下,将样条以10mm/min的拉伸速率拉至300%应变,固
定应变,保持60min。胶料性能见表1。
[0058]
对比例1
[0059]
将6.0g 1,4-苯二硼酸和8.02g 1-硫代甘油溶于200ml四氢呋喃中,加入10.0g无水硫酸镁,在25℃条件下搅拌24小时,过滤,除去无水硫酸镁,旋蒸,得到2,2
’‑
(1,4-亚苯基)-双[4-硫醇1,3,2-二氧硼杂环戊烷](bdb)。
[0060]
将环氧度30%的环氧天然橡胶45g、bdb 4.5g、2-二甲氨基吡啶dmap 0.45g于双辊开炼机上在25℃条件下混合均匀。将混合物剪碎成小颗粒后放入平板流变仪中热压固化,固化温度160℃,固化压力10mpa,固化时间45min。胶料性能见表1。
[0061]
对比例2
[0062]
将6.0g 1,4-苯二硼酸和8.02g 1-硫代甘油溶于200ml四氢呋喃中,加入10.0g无水硫酸镁,在25℃条件下搅拌24小时,过滤,除去无水硫酸镁,旋蒸,得到2,2
’‑
(1,4-亚苯基)-双[4-硫醇1,3,2-二氧硼杂环戊烷](bdb)。
[0063]
将环氧度30%的环氧天然橡胶45g、bdb 2.25g、2-二甲氨基吡啶dmap 0.225g于双辊开炼机上在25℃条件下混合均匀。将混合物剪碎成小颗粒后放入平板流变仪中热压固化,固化温度160℃,固化压力10mpa,固化时间45min。胶料性能见表1。
[0064]
对比例3
[0065]
将6.0g 1,4-苯二硼酸和8.02g 1-硫代甘油溶于200ml四氢呋喃中,加入10.0g无水硫酸镁,在25℃条件下搅拌24小时,过滤,除去无水硫酸镁,旋蒸,得到2,2
’‑
(1,4-亚苯基)-双[4-硫醇1,3,2-二氧硼杂环戊烷](bdb)。
[0066]
将环氧度30%的环氧天然橡胶45g、bdb 0.45g、2-二甲氨基吡啶dmap 0.045g于双辊开炼机上在25℃条件下混合均匀。将混合物剪碎成小颗粒后放入平板流变仪中热压固化,固化温度160℃,固化压力10mpa,固化时间45min。胶料性能见表1。
[0067]
表1本发明实施例1-5及对比例1-3性能对比表
[0068]
[0069]
本发明样品的拉伸强度以及修复后的拉伸强度均采用用美国instron公司万能材料试验机测试,测试样条为长37.5mm,厚1mm,宽2mm的哑铃型样条,拉伸速率为500mm/min,样条的修复拉伸强度是指样条切断后,将断面贴合1min,后放于80℃条件下自行愈合24h后所测。
[0070]
由表1中数据可得,实施例1~3相较于对比例1~3具有更高的拉伸强度,最高可达30mpa以上,在80℃修复24h后,也较对比例1~3的修复样条具有更高的拉伸强度。比较实施例1、4和5,可以发现通过控制不同的松弛时间可以获得不同性能的样品。综上可知,本发明制备了一种高强度的自修复弹性体材料,并提供了一种可行的制备方法。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1