一种刺葡萄VdERF2基因及其编码蛋白和应用
一种刺葡萄vderf2基因及其编码蛋白和应用
技术领域
1.本发明涉及生物技术领域,尤其涉及一种刺葡萄vderf2基因及其编码蛋白和应用。
背景技术:
2.葡萄是世界上重要的经济果树,不仅可鲜食,还被广泛用于酿酒、制汁等。然而,大多数的葡萄品种对各种病原菌高度敏感,其中,由c.viniferum引起的葡萄炭疽病,严重影响葡萄的产量与品质,从而造成重大的经济损失。由于该真菌主要侵染成熟果实,还会影响葡萄酒的化学性质。在温暖潮湿的条件下,葡萄炭疽病在果粒、枝梢、叶片上可见典型的病变症状。
3.目前在葡萄栽培生产中,主要用杀菌剂来控制炭疽病菌,但杀菌剂的使用不仅危害环境与健康,还会增强病原菌的抗药性。由于使用杀菌剂控制炭疽病传播的不良影响,人们越来越关注将欧美杂种与欧亚种葡萄杂交来选育抗炭疽病的鲜食葡萄。近年来葡萄抗炭疽病基因位点(cgr1)鉴定已有相关研究报道,但葡萄感染炭疽病的分子机制目前还不清楚。因此,葡萄抗炭疽病育种是南方葡萄栽培生产中亟需解决的问题。
技术实现要素:
4.本发明要解决的技术问题,在于提供一种刺葡萄vderf2基因及其编码蛋白,可以提高植株对炭疽病的抗性。
5.本发明是这样实现的:
6.本发明首先提供了一种刺葡萄vderf2基因,位于刺葡萄的2号染色体上,分布于177614~179312区域。
7.具体地,所述刺葡萄vderf2基因的核苷酸序列如seqidno.1所示。
8.本发明还提供了所述刺葡萄vderf2基因编码的蛋白质,其氨基酸序列如seqidno.2所示,含有ap2结构域,且与欧洲葡萄同源性最高。
9.以及,含有所述刺葡萄vderf2基因的重组表达载体。
10.进一步地,所述重组表达载体包含所述刺葡萄vderf2基因及与其可操作连接的pcambia2300-gfp载体。
11.最后,本发明提供了所述刺葡萄vderf2基因在提高植物对病原菌的抗性中的应用。病原菌胁迫下vderf2基因表达量升高,激发了sa通路上的pr基因,从而增强对病原菌的抗性。
12.进一步地,所述病原菌为致病炭疽菌。
13.进一步地,所述植物包括番茄。
14.本发明具有如下优点:
15.本发明提供了一种刺葡萄vderf2基因,其核酸序列为seqidno.1所述,全长849bp,编码282个氨基酸,氨基酸序列为seqidno.2所示,含有ap2结构域,且与欧洲葡萄高度同源,
编码的蛋白位于细胞核上。
16.本发明分析了刺葡萄果实经炭疽菌侵染后,其果皮中vderf2的表达情况,实验证明,随侵染的进行,vderf2表达量持续升高,在第7d达到高峰,vderf2可响应葡萄炭疽病而诱导表达。
17.本发明通过克隆vderf2序列,构建过表达载体pcambia2300-vderf2-gfp,转化micro-tom番茄获得转基因番茄,果实接种炭疽菌后,相较于野生型番茄发病症状较轻,且vderf2转基因果实中的slpr1和slpr2基因的表达量在接种炭疽菌后各个时间段均显著升高。进一步验证了vderf2基因过表达可增强植株对炭疽病的抗性。因此,本发明提供的vderf2基因能提高植株对炭疽病抗性,为植物抗炭疽病育种提供理论基础。
附图说明
18.下面参照附图结合实施例对本发明作进一步的说明。
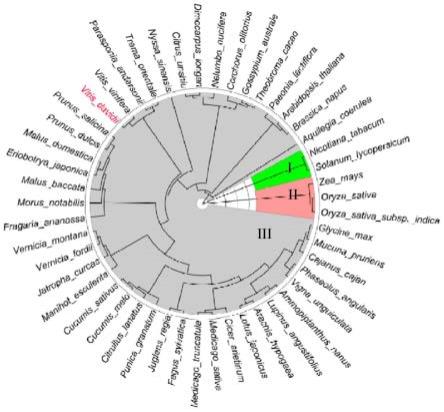
19.图1为刺葡萄与部分物种erf2同源蛋白序列聚类分析,其中红色字体代表刺葡萄(vitis davidii)vderf2,i、ii和iii代表不同类别。
20.图2为vderf2基因构建到pcambia2300-gfp载体上示意图。
21.图3为刺葡萄vderf2蛋白结构域分析图。
22.图4为刺葡萄vderf2染色体定位。
23.图5为vderf2响应葡萄炭疽病诱导表达量分析图。
24.图6为vderf2亚细胞定位分析图(标尺为25μm)。
25.图7为阳性转基因植株pcr检测结果;a:pcr检测转基因番茄;b:qrt-pcr检测转基因番茄;m:dnamaker(d5000);po:阳性对照;wt:野生型。
26.图8为转vderf2基因株系农艺性状图,其中a为转vderf2番茄植株与对照组的植株(左为野生型,右为转基因型),b为转vderf2植株与对照组的果实大小(左为野生型,右为转基因型)。
27.图9为转vderf2基因番茄果实性状指标分析,其中a为转vderf2番茄与对照组的果实单果重比较,b为转vderf2番茄与对照组的果实横、纵径比较。
28.图10为过表达vderf2转基因番茄果实对炭疽菌抗性的分析图,其中a为番茄果实接种炭疽菌72h后表型(左为野生型,右为转基因型);b-c为接种炭疽菌0h、6h、12h、24h、48h和72h后转基因株系和野生型番茄果实slpr1(b)和slpr2(c)的相对表达量。
具体实施方式
29.下面结合附图通过实施例来对本发明进行详细说明,但并不是对本发明的限制,仅作为示例说明。
30.以下实施例中所使用的实验方法如无特殊说明,均为常规方法。
31.以下实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
32.实施例中所用的菌种及质粒:
33.试验所用的过表达载体为pcambia2300-gfp;转基因及烟草表皮细胞定位分析所用的侵染农杆菌为gv3101(p19+psoup)菌株;所用的大肠杆菌top10感受态细胞从tiangen公司购买。
34.实施例中所用的主要试剂包括反转录试剂盒,qrt-pcrsuppermix(transgen公司),质粒提取试剂盒,植物rna提取试剂盒,胶回收试剂盒,la高保真酶,蔗糖,ms粉末,tris,琼脂粉,ctab,75%乙醇,无水乙醇。
35.实施例中番茄转基因过程中的培养基和激素浓度:
36.ms液体培养基:4.43g ms粉末和30g蔗糖溶于1l蒸馏水中。ms固体培养基:4.43g ms粉末和30g蔗糖溶于1l蒸馏水中,同时加入1.5g琼脂粉(ph=6.0,2mol/lnaoh进行调节)。预培养:ms固体培养基+1mg/l kt。分化培养:ms固体培养基灭菌后,55℃加入300mg/l tim+2mg/l zt+50mg/l kan。生根培养:ms固体培养基灭菌后,55℃加入300mg/l tim+1mg/l iaa+50mg/l kan。
37.实施例1vderf2 cds序列克隆与载体构建
38.1.提取rna
39.以刺葡萄果皮为材料,提取rna。
40.2.cdna合成
41.以上述rna为模板,反转录合成cdna,具体操作为:取总rna1μg、oligo dt primer(50μm)及dntps(10mm)各1μl、无rna酶水补齐至20μl,混匀后65℃孵育5min,冰上放置3min,短暂离心。反应组分如表1所示,并将反应液先置于42℃孵育30min;随后70℃孵育15min;4℃孵育10min,最终获得cdna,可用于后续的试验。
42.表1反应液配制
[0043][0044][0045]
3.克隆目的基因vderf2
[0046]
以cdna为模板,用引物p2300-vderf2-bamhi-f seqidno.3和p2300-vderf2-sali-r seqidno.4进行pcr扩增,扩增体系如下:
[0047][0048]
pcr循环反应条件:94℃预变性1min,(94℃变性30s,57℃退火1min,72℃延伸2min)29个循环,72℃终延伸5min。pcr产物使用1%的琼脂糖凝胶电泳检测,目标条带强度用bio imaging systems software(syngene,cambridge,uk)分析。
[0049]
seq id no.3:
[0050]
p2300-vderf2-bamhi-f:
[0051]
tcggtacccggggatccatgtgtgattacagtagtaatcc;
[0052]
seq id no.4:
[0053]
p2300-vderf2-sali-r:
[0054]
gctcaccatggtgtcgacgcgaaccaatagctgtgggccatgtg
[0055]
注:小写序列为接头引物(载体序列),大写为基因序列。
[0056]
4.pcr扩增产物回收
[0057]
pcr产物使用1%琼脂糖凝胶电泳检测,紫外灯下切下单一的电泳条带。回收过程参照试剂盒说明书进行:吸附柱中加入500μl平衡液,12000rpm离心1min弃废液后吸附柱待用。胶块中加入3倍重量溶胶液50℃水浴溶解彻底后,室温静置10min。将溶解液加入准备好的吸附柱中静置1min,12000rpm离心30s,弃废液。吸附柱中加入600μl漂洗液,12000rpm离心30s后弃废液,重复一次。弃掉漂洗液后离心2min,吸附柱开盖5min;加30μl ddh2o室温静置2min,12000rpm离心2min备用。
[0058]
5.构建载体
[0059]
参照博迈德公司的无缝克隆试剂盒进行同源重组反应(如图2),将上述获得的胶回收产物,与线性化载体pcambia2300混合,同时加入反应液,反应体系为10μl,在50℃下反应15min,随后冰浴15min,进行后续的反应。
[0060]
6.转化感受态细胞
[0061]
上述重组反应液中,加入25μl的top10感受态细胞,轻弹混匀。冰浴20min,42℃热激90s,冰浴5min,加入300μl液体lb,37℃复苏30min,随后涂布在50mg
·
l-1
kan平板上,37℃过夜培养进行阳性克隆筛选。
[0062]
7.重组克隆载体鉴定
[0063]
将1ml附加50mg
·
l-1
kan lb液体培养基装入2ml离心管中,从lb平板上挑选单菌落接种于其中,37℃振荡过夜培养。以过夜培养的菌液为模板,进行pcr扩增,扩增体系如下:
[0064][0065]
pcr扩增条件为:94℃1min,(94℃30s,57℃1min,72℃2min)31个循环,72℃5min。使用1%琼脂糖凝胶电泳检测pcr产物,在凝胶成像系统上确认正确的目标条带,将正确的菌液在10m液体lb(附加50mg
·
l-1
kan)中37℃振荡培养用于后续试验。
[0066]
8.重组质粒提取
[0067]
上述培养好的菌液取500μl用于测序,另取500μl菌液与30%甘油1:1混匀,-80℃保存备用。余下的质粒进行质粒提取,取2ml菌液,12000rpm离心1min;菌体重悬于250μl溶液i;加入250μl溶液ii,轻柔混匀,室温静置2min;加入350μl溶液iii,轻柔混匀;12000rpm离心10min;取上清液,过吸附柱,12000rpm离心1min;加漂洗液700μl过柱清洗两次,弃掉漂洗液后离心2min,吸附柱开盖5min;加50μlddh2o室温静置2min,12000rpm离心2min,-20℃保存。
[0068]
9.酶切分析
[0069]
取1μg上述质粒用于酶切检测,反应体系为20μl:10
×
enzyme buffer2μl,bamh i/sal i各0.5μl,用ddh2o补至20μl,37℃反应3h。使用1%凝胶电泳检测酶切产物,将酶切正确的质粒用于测序,并最终确认正确的质粒用于后续试验。
[0070]
实施例2vderf2生物信息学分析
[0071]
将vderf2的氨基酸序列放到blast网站上进行序列比对,并将不同物种的同源序列下载,用于构建系统进化树。vderf2的氨基酸序列相似性,使用dnaman(v6.0)进行多重比较。使用gendoc(v2.7.0)软件输出序列多重比较分析结果。vderf2基因在染色体上的位置预测在葡萄基因组网站genoscope genome browser中分析。结果如图4,所克隆的vderf2位于2号染色体上,分布于177614-179312的区域。系统进化树的构建在phylogeny.frplatform(http://www.phylogeny.fr)上,采用最大似然法(maximum likelihood)进行。
[0072]
以vderf2氨基酸序列应用clustw程序与欧洲葡萄、拟南芥、栽培稻、烟草、番茄和玉米进行了比对。结果如图3所示,vderf2长849bp,编码282个氨基酸,与其他物种比对后发现均含有ap2结构域(137aa-200aa)。将刺葡萄中的vderf2氨基酸序列与genbank中搜索的其他属植物erf氨基酸序列进行同源性比较,应用phylogeny.frplatform中的最大似然法构建进化树,如图1所示。从刺葡萄中克隆的vderf2基因与欧洲葡萄聚为一小簇,表明与欧洲葡萄关系最近。
[0073]
ap2/erf转录因子超家族的共同特征是都具有保守的ap2/erf结构域。根据ap2/erf结构域的个数以及是否含有其他的结构域,将ap2/erf转录因子超家族分为三个家族:ap2家族(含有两个重复的ap2/erf结构域)、erf家族(只含有一个ap2/erf结构域)、rav家族(除了含有一个ap2/erf结构域以外,还有另外一个b3结构域)。另外,根据ap2/erf结构域保守氨基酸的不同,又将erf转录因子家族分为erf亚家族和cbf/dreb亚家族。erf2为erf家族中erf亚家族成员。
[0074]
实施例3vderf2基因的表达模式分析
[0075]
采用针刺法对刺葡萄进行炭疽菌接种,并采集受炭疽菌侵染后的葡萄果皮为试验材料。根据vderf2基因的序列设计定量引物,qpcr-erf2-f:aacggctcaaactggaatcg;qpcr-erf2-r:tagctgtgggccatgtgtaa。进行实时荧光定量pcr并对vderf2基因的表达模式进行分析。
[0076]
结果如图5所示,刺葡萄接种葡萄炭疽菌后的0d、1d、3d、7d果皮中的vderf2表达量随着侵染的进行,持续升高,在第7d达到高峰。表明vderf2可以响应葡萄炭疽病而诱导表达。
[0077]
实施例4烟草表皮细胞定位分析
[0078]
将测序正确的重组质粒使用电激法,转入农杆菌gv3101中,在含有50mg
·
l-1
kan、50mg
·
l-1
gent和50mg
·
l-1
rif的抗性平板上28℃培养2d。挑取菌落在5ml lb液体培养基中(含有50mg
·
l-1
kan、50mg
·
l-1
gent和50mg
·
l-1
rif,)在28℃下180rpm摇菌16-18h,进行pcr检测。将正确的菌液在含有同样抗生素的10ml lb液体培养基中扩大培养。5000rpm离心5min,弃去上清液,用重悬液(mes 2.130g/l+mgcl 22.03g/l+蔗糖20g/l)重悬菌体并清洗3次。将重悬菌液稀释至od
600
=0.4,随后加入as(200mmol
·
l-1
)避光条室温静置3h激活农杆菌。用无针头的1ml注射器在健康的本氏烟草叶背面注射入重悬菌液,在光照培养箱中培养72h,通过激光共聚焦显微镜下(莱卡tcssp8)观察烟草叶片中gfp融合蛋白的分布,并保存图片。结果如图6所示,表明vderf2-gfp蛋白定位在细胞核上。
[0079]
实施例5过表达载体转化番茄
[0080]
1.番茄无菌苗的获得
[0081]
无菌操作台中将番茄种子(micro-tom)无菌水清洗5次后浸泡30min,70%乙醇浸洗1min,无菌水浸洗3次。然后用10%naclo浸洗10min,无菌水冲洗5次。用无菌滤纸吸干种子上的水分,随后接种到ms固体培养基上,在培养箱25℃暗培养4-5d。光照25℃,16h/黑暗16℃,8h。
[0082]
2.农杆菌侵染和共培养
[0083]
待番茄长出两片子叶且刚平展开时,剪取幼苗子叶,将叶片切成正方形,在ms分化培养基上铺一张灭菌滤纸,将子叶背面向上平铺在培养基上,并放置在光照培养箱中过夜培养,光照25℃,16h/黑暗16℃,8h。
[0084]
将含有重组质粒的农杆菌(gv3101)菌液按1:250加入lb培养基中进行活化,28℃,180rpm培养48h。用2个2ml离心管各吸取2ml活化好的菌液,5000rpm离心5min后,弃上清液,加入1ml ms液体培养基(含50μm as)将菌体充分悬浮后;在90mm培养皿中倒入三分之一高度的ms液体培养基。将悬浮好的2管菌液加入培养皿,后将准备好的番茄叶盘放入侵染液中5min,期间不停摇动培养皿,使菌体充分与子叶盘结合。时间结束后快速吸出侵染液,并用
滤纸吸干叶片表面残余菌液,后将叶片平铺在ms培养基上,封口后放在25℃黑暗培养箱中,培养2d。
[0085]
3.脱菌与分化培养
[0086]
暗培养后的叶盘使用含有抗生素(300mg/ltim)的无菌水进行脱菌,将暗培养后的叶盘洗5遍,无菌滤纸吸去多余的水分。放入分化培养基中,光照25℃,16h/黑暗16℃,8h。
[0087]
分化培养期间观察叶盘分化情况,至叶片伤口处形成愈伤组织并分化出幼苗,将幼苗转移到ms分化培养基中,待幼苗长大,将幼苗切下插入到生根培养基中。
[0088]
4.生根培养与成苗
[0089]
在生根培养基中培养3周左右后,选择根系发育良好的番茄苗,从组培瓶中移出,放入带有透明盖子的营养钵中进行炼苗,炼苗2周,期间逐渐揭开盖子。成苗后的番茄经过检测后,即可进行后续试验。
[0090]
5.转基因番茄的检测
[0091]
取成苗后的t0代番茄幼嫩叶片0.5g,采用ctab法提取基因组dna。以基因组dna为模板,使用pcambia2300载体通用引物(p2300-f:tccttcgcaagacccttcctctat;p2300-r:cagggtcagcttgccgtag)检测vderf2是否插入。经过pcr检测为阳性的植株,选取植株幼嫩叶片,提取总rna,经过反转录后得到cdna,以slactin(slactinf:attccctgactgtttgctagt;slactinr:tccaacacaataccggtggt)为内参引物和vderf2定量引物(qpcr-erf2-f:aacggctcaaactggaatcg;qpcr-erf2-r:tagctgtgggccatgtgtaa)进行半定量分析。
[0092]
结果如下(如图7所示):利用农杆菌介导的植物转化方法将vderf2转化番茄,将移栽成苗的番茄炼苗后,取植株幼嫩叶片提取dna,使用pcambia2300载体的通用引物进行pcr检测。以pcambia2300-vderf2-gfp质粒为阳性对照,野生型番茄植株叶片dna为阴性对照,pcr电泳结果表明候选植株(#12、#13、#16、#17)与阳性对照有一致的条带,说明候选植株中含有vderf2基因,野生型植株中检测不到目的条带。为了进一步验证vderf2基因在上述候选植株中是否表达,研究中提取了候选植株幼嫩叶片的rna并进行反转录,根据vderf2基因的序列设计特异引物并以番茄slactin为内参进行pcr半定量检测,经过28个循环的pcr检测,#12和#16植株中检测到了目的条带,而野生型植株中没有条带,可以鉴定#12和#16植株为阳性植株。结果表明,vderf2基因在阳性植株(#12、#16)中表达。
[0093]
对获得的转基因植株进行表型观察(如图8、图9),野生型番茄植株与vderf2过表达植株在表型上并无明显差别,且野生型番茄与vderf2过表达番茄#12和#16植株在番茄果实的单果重、横径和纵径的长度上没有明显差异。
[0094]
实施例6转基因番茄抗炭疽病鉴定
[0095]
采取成熟、无病斑的番茄果实,使用75%乙醇溶液表面消毒15s,无菌水清洗3遍。番茄尖胞炭疽病菌(c.acutatum)由本实验室保存。接种前,首先使用解剖针在果实顶部造伤处理,使用打孔器打下6mm的pda菌块(预培养7d),接种到造伤部位。同时接种野生型(对照)和转基因果实,在28℃培养箱进行保湿暗培养,48h后移出菌块,于接种后的0h、6h、12h、24h、48h、72h采集果实果皮,液氮速冻,-80℃冰箱保存备用。提取上述样品rna并反转录为cdna(100ng/μl),以番茄slactin为内参,采用qrt-pcr的方法,比较番茄在接种炭疽病后,番茄的slpr1(登录号:eu589238,slpr1-f:ataaagtgatcgattgtcgagga;slpr1-r:taagctgcaacatacacacatcc)和slpr2(登录号:eu589238,slpr2-f:
tctgtagacatgacgttgattgg;slpr2-r:agagcatacggaagtgaaatctg)在不同时间段的表达量。
[0096]
如图10所示,接种vderf2转基因番茄果实和野生型番茄果实72h后发现,vderf2转基因番茄果实接种后,相对于野生型番茄果实中的病斑直径变小,发病症状较轻。进一步地,将野生型番茄果实和vderf2转基因番茄果实接种炭疽菌,并于接种后0h、6h、12h、24h、48h和72h采集番茄果皮,提取rna反转录后,以slactin为内参,实时荧光定量pcr分析了vderf2转基因番茄果实接种炭疽菌后不同时间段,slpr1和slpr2基因的表达量变化。结果表明,相对于野生型番茄果实,vderf2转基因番茄果实中slpr1和slpr2基因的表达量在接种炭疽菌后各个时间段均显著升高,表明过表达vderf2的番茄果实对炭疽菌的抗性提高。
[0097]
虽然以上描述了本发明的具体实施方式,但是熟悉本技术领域的技术人员应当理解,我们所描述的具体的实施例只是说明性的,而不是用于对本发明的范围的限定,熟悉本领域的技术人员在依照本发明的精神所作的等效的修饰以及变化,都应当涵盖在本发明的权利要求所保护的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1