一种双氰乙基叔胺的加氢方法与流程
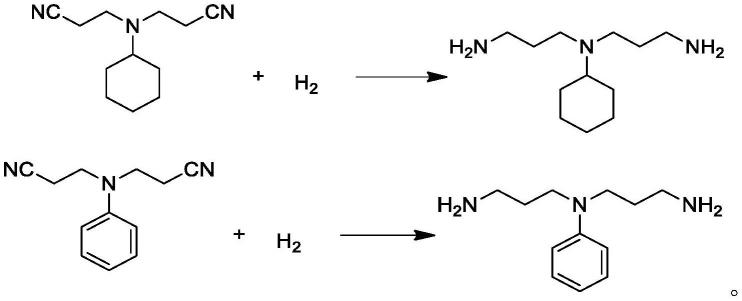
1.本发明属于有机合成领域,具体涉及一种双氰乙基叔胺的加氢方法。
背景技术:
2.相较于传统的胺类固化剂,氰乙基胺类固化剂在硬度、韧性以及凝胶速度等方面有着更加显著的优势,有望被应用在高端环氧固化剂领域。利用氰乙基化方式得到的改性胺通常有单氰乙基仲胺和双氰乙基叔胺两种结构,其中叔胺的固化效果一般更加柔顺性更好、抗冲击性更强;而为了能够提升双氰乙基叔胺的胺值,现有的技术手段是采用加氢的方式将双氰乙基加氢得到双氨丙基,从而进一步改善氰乙基叔胺活性差这一问题,进而满足更加高端的环氧固化领域。
3.相较于单氰乙基仲胺类的合成,制备双氰乙基叔胺的工艺更加复杂,通常需要添加一定量的酸进行催化合成,这主要是由于双氰乙基叔胺上的氮原子空间位阻过大的缘故导致;然而这就会进一步影响到后续加氢步骤的进行,酸对于加氢催化剂的毒害作用非常强,而且由于氢解以及缩合等一列的副反应导致的游离胺增加,会进一步与贵金属催化剂进行缩合,这就进一步加快了催化剂失活速率加快、使用寿命严重不足,所以研发难度极大,在现有的公开技术中,一直没有设计或解决相应问题的办法,严重地阻碍了改性胺类固化剂的发展。
4.cn 113372241 a公开了一种双氰乙基叔胺的合成方法,该方法是以乙醇酸为催化剂合成双氰乙基叔胺,但并未进一步介绍双氰乙基叔胺的加氢工艺以及乙醇酸对于加氢阶段的影响,缺乏借鉴意义。
5.现有技术中涉及到腈加氢催化剂的相关专利主要有:cn 2011110070427.5公开了一种间歇高压釜加氢制备间苯二甲胺的工艺,该发明所采用的骨架镍催化剂只可以套用10个批次。公开号为cn 112047843 a的专利公布了一种提高间苯二甲胺催化剂固定床加氢催化剂稳定性的方法,在发明在加氢催化剂与原料之间设置保护剂,从而有效去除反应过程中的有害物质对催化剂的毒害,延长了加氢催化剂的使用寿命。cn 108276291 a提供了一种腈加氢催化剂的制备方法,该发明为了提高加氢催化剂的使用寿命,采用共沉淀法制备了相应的coni合金催化剂。另外cn 108393092 b同样了一种腈类化合物加氢制备仲胺的催化剂制备方法,该工艺采用浸渍法、共沉淀法、化学气相沉积法、原子层沉积法等制备了不易失活的加氢催化剂。可见腈加氢催化剂存在严重的失活现象,而且普遍针对催化剂失活的问题开展了大量的研究,但研究范围较窄,不具有代表性。
6.综上所述,腈加氢的现有技术依然存在较多缺陷,而且双氰乙基叔胺的合成以及加氢工艺更为复杂,至今未建立针对性的研究体系,这严重影响了改性胺类在高端环氧树脂固化剂领域的应用。
技术实现要素:
7.本发明的目的在于提供一种双氰乙基叔胺的加氢方法。本发明仅通过工艺改进就
实现了催化剂套用寿命的延长,在保持产品收率不变的同时降低了催化剂单耗,而且很大程度上也减少了双氰乙基叔胺原料的预处理脱酸步骤,既提升了生产效率,也降低了生产成本,非常适用于氰乙基胺化合物的加氢工业化生产。
8.为了实现上述目的,本发明提供了如下技术方案:
9.一种双氰乙基叔胺的加氢方法,所述方法包含如下步骤:
10.s1:以游离胺吸附剂、加氢催化剂和有机溶剂铺底,通入h2,以已预处理脱酸的双氰乙基叔胺为原料进行加氢反应;
11.s2:切换未预处理脱酸的双氰乙基叔胺为原料继续加氢反应;
12.s3:蒸馏提纯反应母液,得到目标产品。
13.本发明通过添加游离胺吸附剂解决了由于氢解或者缩聚等副反应所造成催化剂活性位点减少的问题;进一步以切换已预处理和未处理脱酸双氰乙基叔胺两种原料先后进料的方式,通过酸对游离胺吸附剂的去保护作用,实现了游离胺吸附剂络合副产物的分解,减轻了双氨丙基反应母液后处理的工艺难度。本工艺不仅延长了催化剂套用寿命,在保持产品收率不变的同时降低了催化剂单耗。
14.示意性地,部分加氢反应涉及的反应式如下:
[0015][0016]
本发明中,s1所述游离胺吸附剂为触酸分解型吸附剂,优选三苯基氯甲烷(trtcl)、氯甲酸苄酯(cbzcl)、2,4-二甲氧基苯甲醛、苯甲醛、二碳酸二叔丁酯(boc2o)中的一种或多种,优选氯甲酸苄酯(cbzcl)和/或二碳酸二叔丁酯(boc2o);优选地,游离胺吸附剂的用量为预处理脱酸的双氰乙基叔胺原料质量的2~15wt%,优选5~8wt%。
[0017]
本发明中,s1所述加氢催化剂为雷尼镍、雷尼钴、雷尼铜、雷尼锌、负载镍催化剂、负载钴催化剂中的一种或多种,优选雷尼镍和/或雷尼钴;优选地,催化剂的用量为双氰乙基叔胺总质量的1~25wt%,优选5~15wt%。
[0018]
本发明中,s1所述溶剂为甲醇、乙醇、异丙醇、二氧六环、四氢呋喃、苯、甲苯中的一种或多种,优选甲醇和/或乙醇;优选地,溶剂的用量为双氰乙基叔胺总质量的1~8倍,优选4~6倍。
[0019]
本发明中,s1所述的双氰乙基叔胺为双氰乙基环戊胺、2-甲基-双氰乙基环戊胺、双氰乙基环己胺、双氰乙基苯胺、2-甲基-双氰乙基环己胺、2-甲基-双氰乙基苯胺、2,3-二甲基-双氰乙基环己胺、2,3-二甲基-双氰乙基苯胺中的一种或多种,优选双氰乙基环己胺和/或双氰乙基苯胺;优选地,s1所述已预处理脱酸的双氰乙基叔胺原料包含伯胺0.5~2wt%,单氰乙基仲胺1~2wt%,双氰乙基叔胺96~98.5wt%,以已预处理脱酸的双氰乙基叔胺原料总质量计。
[0020]
本发明中,s1所述双氰乙基叔胺原料中的酸为盐酸、硫酸、磷酸、乙醇酸、醋酸、草酸中的一种或多种,优选磷酸和/或草酸;优选地,s1所述已预处理脱酸的双氰乙基叔胺原料中酸含量为20~200ppm,优选50~150ppm,以已预处理脱酸的双氰乙基叔胺原料总质量计;优选地,获得s1所述已预处理脱酸的双氰乙基叔胺原料的处理方法为碱液洗涤,优选采用na2co3、k2co3、(nh4)2co3、lioh、naoh、koh中的一种或多种的水溶液洗涤,更优选采用(nh4)2co3和/或lioh水溶液洗涤。
[0021]
本发明中,s1所述加氢反应的反应温度为50~150℃,优选70~130℃;h2压力为绝压1~10mpa,优选绝压3~8mpa;进料结束后反应延长时间为0.5~5min,优选0.5~2min。
[0022]
本发明中,s2所述的未预处理脱酸的双氰乙基叔胺原料中酸含量为0.5~10wt%,优选1~6wt%,以未预处理脱酸的双氰乙基叔胺原料总质量计;优选地,s1已预处理脱酸的双氰乙基叔胺原料与s2未预处理脱酸的双氰乙基叔胺原料的质量比为1:1~10,优选1:3~5;优选地,s2所述的加氢反应的反应温度为50~150℃,优选70~130℃;h2压力为绝压1~10mpa,优选3~8mpa;进料结束后反应延长时间为0.5~5h,优选0.5~2.5h。
[0023]
本发明中,所述s3蒸馏提纯的温度为50~120℃,优选为80~100℃;绝对压力为1~5kpa,优选2~3kpa,处理时长为1~5h,优选2~3h。
[0024]
本发明的另一目的在于提供一种双氰乙基叔胺的加氢产品。
[0025]
一种双氰乙基叔胺的加氢产品,采用上述的加氢方法制备获得,所述产品为双氨丙基环戊胺、2-甲基-双氨丙基环戊胺、双氨丙基环己胺、双氨丙基苯胺、2-甲基-双氨丙基环己胺、2-甲基-双氨丙基苯胺、2,3-二甲基-双氨丙基环己胺、2,3-二甲基-双氨丙基苯胺中的一种或多种,优选双氨丙基环己胺和/或双氨丙基苯胺。
[0026]
在一种实施方案中,所述产品中氨丙基脂环仲胺的含量为0-3%,双氨丙基脂环叔胺含量为95-98%,其它产物0-2%,以产品总质量计。
[0027]
本发明的另一目的在于提供一种双氰乙基叔胺的加氢方法的用途。
[0028]
一种双氰乙基叔胺的加氢方法的用途,所述方法为上述的加氢方法,所述方法用于双氰乙基叔胺加氢制备双氨丙基环戊胺、2-甲基-双氨丙基环戊胺、双氨丙基环己胺、双氨丙基苯胺、2-甲基-双氨丙基环己胺、2-甲基-双氨丙基苯胺、2,3-二甲基-双氨丙基环己胺、2,3-二甲基-双氨丙基苯胺中的一种或多种,优选用于双氰乙基叔胺加氢制备双氨丙基环己胺和/或双氨丙基苯胺。
[0029]
本发明技术方案的有益效果在于:
[0030]
(1)延长了催化剂套用寿命,产品平均收率≥95%,催化剂单耗最低下降至1.5kg/t。
[0031]
(2)很大程度上也减少了双氰乙基叔胺原料的预处理脱酸步骤,提升了生产效率,也降低了生产成本。
具体实施方式
[0032]
下面结合实施例,对本发明作出进一步的说明,但本发明不限于所列出的实施例,还应包括在本发明所附的权利要求书中的技术方案的有效改进和延伸。
[0033]
主要原料的来源:
[0034]
催化剂:雷尼钴、雷尼镍,万华化学;
[0035]
预处理脱酸双氰乙基环己胺,万华化学:其中包括环己胺0.6wt%,单氰乙基环己胺1.5wt%,双氰乙基环己胺97.9wt%,以原料双氰乙基环己胺总量计;脱酸方法采用(nh4)2co3水溶液洗涤法,已预处理脱酸的双氰乙基环己胺原料中酸含量为200ppm,以已预处理脱酸的双氰乙基环己胺原料总质量计;
[0036]
未预处理脱酸双氰乙基环己胺,万华化学:其中包括环己胺0.6wt%,单氰乙基环己胺1.5wt%,双氰乙基环己胺97.9wt%,以原料双氰乙基环己胺总量计;未预处理脱酸的双氰乙基环己胺原料中酸含量为6wt%,以未处理脱酸的双氰乙基环己胺原料总质量计;
[0037]
预处理脱酸双氰乙基苯胺,万华化学:其中包括苯胺1.0wt%,单氰乙基苯胺1.8wt%,双氰乙基苯胺97.2wt%,以原料双氰乙基苯胺总量计;脱酸方法采用k2co3水溶液洗涤法,已预处理脱酸的双氰乙基苯胺原料中酸含量为100ppm,以已预处理脱酸的双氰乙基苯胺原料总质量计;
[0038]
未预处理脱酸双氰乙基苯胺,万华化学:其中包括苯胺1.0wt%,单氰乙基苯胺1.8wt%,双氰乙基苯胺97.2wt%,以原料双氰乙基苯胺总量计;未预处理脱酸的双氰乙基苯胺原料中酸含量为1wt%,以未处理脱酸的双氰乙基苯胺原料总质量计;
[0039]
氯甲酸苄酯(cbzcl):纯度96%,阿拉丁;
[0040]
二碳酸二叔丁酯(boc2o):纯度99%,阿拉丁;
[0041]
甲醇:纯度99.5%,阿拉丁;
[0042]
乙醇:纯度99.5%,阿拉丁;
[0043]
盐酸:纯度37%,阿拉丁。
[0044]
测试方法:
[0045]
气相色谱:采用安捷伦7890和db-5(30mm
×
0.25mmid
×
0.25μm),进样器温度为280℃,检测器温度为300℃。升温程序如下:起始柱温为50℃,保持2min;以5℃/min升温至80℃,保持0min;以15℃/min升温至300℃,保持15min。通过归一化法测定组分含量。
[0046]
酸测试方法:采用瑞士万通电位滴定仪;称取10g样品,加入100ml甲醇,搅拌溶解后,在电位滴定仪上,以非水相酸碱电极为指示电极,以0.02mol/l氢氧化钾-甲醇标准溶液滴定。
[0047]
实施例1
[0048]
s1:采取釜式半间歇工艺,在反应釜中添加12.3g雷尼钴催化剂、10.3g氯甲酸苄酯(cbzcl)以及820g甲醇溶剂,在升温至130℃下将h2绝对压力补充至8mpa,继续将205g已经预处理脱酸的双氰乙基环己胺原料添加进入反应釜中,开始进行加氢反应制备双氨丙基环己胺;
[0049]
s2:当步骤s1中经预处理脱酸的双氰乙基环己胺进料结束之后2min,将原料切换为未预处理脱酸的双氰乙基环己胺,在保持温度130℃、h2绝对压力8mpa的条件下,继续将615.5g未处理脱酸的双氰乙基环己胺原料添加进入反应釜中,并延长反应时间0.5h至吸氢结束;
[0050]
s3:当步骤s2反应停止后,收集反应母液,在80℃,绝对压力3kpa条件下通过蒸馏脱轻组分2h,即可得到纯品双氨丙基环己胺;产品由色谱进行分析结果;重新开始上述s1至s3的操作,一直套用催化剂,至双氨丙基环己胺收率下降至95%以下即停止,套用结果见表1。
[0051]
表1
[0052][0053][0054]
实施例2
[0055]
s1:采取釜式半间歇工艺,在反应釜中添加29.9g雷尼镍催化剂、15.9g二碳酸二叔丁酯(boc2o)以及1194g乙醇溶剂,在升温至70℃下将h2绝对压力补充至3mpa,继续将199g已经预处理脱酸的双氰乙基苯胺原料添加进入反应釜中,开始进行加氢反应制备双氨丙基苯胺;
[0056]
s2:当步骤s1中经预处理脱酸的双氰乙基苯胺进料结束之后1min,将原料切换为未预处理脱酸的双氰乙基苯胺,在保持温度70℃、h2绝对压力3mpa的条件下,继续将995g未处理脱酸的双氰乙基苯胺原料添加进入反应釜中,延长反应时间2.5h至吸氢结束;
[0057]
s3:当步骤s2反应停止后,收集反应母液,在100℃,绝对压力2kpa条件下通过蒸馏脱轻组分3h,即可得到纯品双氨丙基苯胺;产品由色谱进行分析结果;重新开始上述s1至s3的操作一直套用,至双氨丙基苯胺收率下降至95%以下即停止,套用结果见表2。
[0058]
表2
[0059][0060][0061]
实施例3
[0062]
s1:采取釜式半间歇工艺,在反应釜中添加16.4g雷尼钴催化剂、12.3g二碳酸二叔丁酯(boc2o)以及1025g乙醇溶剂,在升温至90℃下将h2绝对压力补充至4mpa,继续将205g已经预处理脱酸的双氰乙基环己胺原料添加进入反应釜中,开始进行加氢反应制备双氨丙基环己胺;
[0063]
s2:当步骤s1中经预处理脱酸的双氰乙基环己胺进料结束之后1min,将原料切换
为未预处理脱酸的双氰乙基环己胺,在保持温度90℃、h2绝对压力4mpa的条件下,继续将820g未处理脱酸的双氰乙基环己胺原料添加进入反应釜中,并延长反应时间2h至吸氢结束;
[0064]
s3:当步骤s2反应停止后,收集反应母液,在90℃,绝对压力2kpa条件下通过蒸馏脱轻组分2h,即可得到纯品双氨丙基环己胺;产品由色谱进行分析结果;重新开始上述s1至s3的操作,一直套用催化剂,至双氨丙基环己胺收率下降至95%以下即停止,套用结果见表3。
[0065]
表3
[0066][0067][0068]
实施例4
[0069]
s1:采取釜式半间歇工艺,在反应釜中添加23.9g雷尼钴催化剂、13.9g氯甲酸苄酯(cbzcl)以及796g甲醇溶剂,在升温至110℃下将h2绝对压力补充至6mpa,继续将199g已经预处理脱酸的双氰乙基苯胺原料添加进入反应釜中,开始进行加氢反应制备双氨丙基苯胺;
[0070]
s2:当步骤s1中经预处理脱酸的双氰乙基苯胺进料结束之后1.5min,将原料切换为未预处理脱酸的双氰乙基苯胺,在保持温度110℃、h2绝对压力6mpa的条件下,继续将995g未处理脱酸的双氰乙基苯胺原料添加进入反应釜中,延长反应时间1h至吸氢结束;
[0071]
s3:当步骤s2反应停止后,收集反应母液,在90℃,绝对压力2kpa条件下通过蒸馏脱轻组分2h,即可得到纯品双氨丙基苯胺;产品由色谱进行分析结果;重新开始上述s1至s3的操作一直套用,至双氨丙基苯胺收率下降至95%以下即停止,套用结果见表4。
[0072]
表4
[0073]
[0074][0075]
对比例1
[0076]
参照实施例1,区别在于不添加游离胺吸附剂。
[0077]
s1:采取釜式半间歇工艺,在反应釜中添加12.3g雷尼钴催化剂以及820g甲醇溶剂,在升温至130℃下将h2绝对压力补充至8mpa,继续将205g已经预处理脱酸的双氰乙基环己胺原料添加进入反应釜中,开始进行加氢反应制备双氨丙基环己胺;
[0078]
s2:当步骤s1中经预处理脱酸的双氰乙基环己胺进料结束之后2min,将原料切换为未预处理脱酸的双氰乙基环己胺,在保持温度130℃、h2绝对压力8mpa的条件下,继续将615.5g未处理脱酸的双氰乙基环己胺原料添加进入反应釜中,并延长反应时间0.5h至吸氢结束;
[0079]
s3:当步骤s2反应停止后,收集反应母液,在80℃,绝对压力3kpa条件下通过蒸馏脱轻组分2h,即可得到纯品双氨丙基环己胺;产品由色谱进行分析结果;重新开始上述s1至s3的操作,一直套用催化剂,至双氨丙基环己胺收率下降至95%以下即停止,套用结果见表5。
[0080]
表5
[0081][0082]
对比例2
[0083]
参照实施例2,区别在于不切换未脱酸的双氰乙基苯胺原料。
[0084]
s1:采取釜式半间歇工艺,在反应釜中添加29.9g雷尼镍催化剂、79.6g二碳酸二叔丁酯(boc2o)以及1194g乙醇溶剂,在升温至70℃下将h2绝对压力补充至3mpa,继续将995g已经预处理脱酸的双氰乙基苯胺原料添加进入反应釜中,开始进行加氢反应制备双氨丙基苯胺,进料结束后延长反应时间1.5h至吸氢结束;
[0085]
s3:当步骤s1反应停止后,收集反应母液,在室温下添加20g盐酸进行搅拌1h,继续在100℃、绝对压力2kpa条件下通过蒸馏脱轻组分3h,即可得到纯品双氨丙基苯胺;产品由色谱进行分析结果;重新开始上述s1、s3的操作一直套用,至双氨丙基苯胺收率下降至95%以下即停止,套用结果见表6。
[0086]
表6
[0087][0088]
对比例3
[0089]
参照实施例3,区别在于只投用未脱酸的双氰乙基环己胺原料,而且不添加游离胺吸附剂。
[0090]
s1:采取釜式半间歇工艺,在反应釜中添加16.4g雷尼钴催化剂以及820g乙醇溶剂,在升温至90℃下将h2绝对压力补充至4mpa,继续将1025g已经未处理脱酸的双氰乙基环己胺原料添加进入反应釜中,开始进行加氢反应制备双氨丙基环己胺;待进料结束后,延长反应时间2h至吸氢结束;
[0091]
s3:当步骤s1反应停止后,收集反应母液,在90℃,绝对压力2kpa条件下通过蒸馏脱轻组分2h,即可得到纯品双氨丙基环己胺;产品由色谱进行分析结果;重新开始上述s1、s3的操作,一直套用催化剂,至双氨丙基环己胺收率下降至95%以下即停止,套用结果见表7。
[0092]
表7
[0093][0094]
本发明的应用不限于以上的实施例,对于本领域技术人员来说,凡在本发明精神下所作相关任何修饰或变更,皆包括在本发明意图保护之范畴。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1