一种谷氨酸棒杆菌高效突变体及重组菌构建方法与应用
1.本发明属于基因工程和诱变技术领域,尤其涉及一种谷氨酸棒杆菌高效突变体及重组菌构建方法与应用。
背景技术:
2.目前针对谷氨酸棒杆菌(c.glutamicum)的突变方法还是比较传统的诱变方法,如紫外诱变、artp诱变、化学诱变等,比较费时费力,且突变频率不高。紫外诱变能够造成基因组随机突变,操作简单,但是有一定的序列偏好性,突变效率较低,且诱变的过程中的紫外线容易对实验人员的皮肤产生损伤。相对于紫外诱变,artp诱变具有更高的突变率和更广的突变谱,且操作简单,但是artp诱变需要消耗大量的活性粒子(如氦粒子),耗费能量。化学诱变剂如甲基磺酸乙酯和1-甲基-3-硝基-1-亚硝基胍,可以用来实现突变体文库的构建以及菌株选育,但是化学诱变产生的突变谱比较狭窄,而且使用的化学突变试剂通常都是剧毒性物质、强有力的人类致癌物。碱基突变类型一般具有12种,通过更多的覆盖12种突变类型,能够赋予突变子所获得的的氨基酸具有更多的突变方向,也就说更广的突变谱能够促进进化的遗传多样性。常规的实验室进化突变方法都存在一定的弊端,因此,寻求突变条件更加温和、环保、健康和安全以及具备更高突变效率和突变谱的随机突变体系引发了人们的广泛关注。
3.哈佛大学david liu团队开发了一种全新的突变质粒mp6,在阿拉伯糖诱导的pbad启动子的调控下,表达了dna聚合酶dnaq926、dna甲基化酶(dam)、半甲基化gatc结合域(seqa)、尿嘧啶dna糖基化酶抑制剂ugi、胞嘧啶脱氨酶(cdai)和细胞内dntp池转录抑制因子(emrr),导致染色体突变率比野生型大肠杆菌提高了322,000倍。相对于xl1-red和endoms/nucs突变系统,mp6突变质粒具有更高的突变率以及更广的突变谱,突变率高达μbp=6.2
×
10-6
。
4.大肠杆菌体系虽然具备了高效突变体系mp6,但是实验室前期研究发现,mp6突变质粒基本不能够在谷氨酸棒杆菌发挥其高效的诱变能力,因此,构建一种谷氨酸棒杆菌突变体系并将其应用于氨基酸生产中是亟待解决的技术问题。
技术实现要素:
5.有鉴于此,本发明的目的在于提供了一种谷氨酸棒杆菌高效突变体及重组菌构建方法与应用。
6.为了实现上述发明目的,本发明提供了以下技术方案:
7.本发明提供了一种谷氨酸棒杆菌高效突变体,所述突变体包括mp1~mp5和mp5t中的任意一种:
8.所述mp1包括dna聚合酶cgl1289m,所述dna聚合酶cgl1289m的氨基酸序列如seq id no:13所示;
9.所述mp2包括如下元件:所述cgl1289m和尿嘧啶dna糖基化酶抑制剂ugi;所述
cgl1289m的下游串联ugi,所述ugi的氨基酸序列如seq id no:25所示;
10.所述mp3包括如下元件:所述cgl1289m、ugi和胞嘧啶脱氨酶pmcda1;所述cgl1289m、ugi和pmcda1依次串联;所述pmcda1的氨基酸序列如seq id no:27所示;
11.所述mp4包括如下元件:所述cgl1289m、ugi和腺嘌呤脱氨酶tada-abe8e;所述cgl1289m、ugi和tada-abe8e依次串联,所述tada-abe8e的氨基酸序列如seq id no:29所示。
12.所述mp5包括如下元件:所述cgl1289m、ugi、pmcda1和tada-abe8e;所述cgl1289m、ugi、pmcda1和tada-abe8e依次串联;
13.所述mp5t包括如下元件:温度敏感型复制起始位点repa101、所述cgl1289m、ugi、pmcda1和tada-abe8e;所述repa101、cgl1289m、ugi、pmcda1和tada-abe8e依次串联,所述repa101的核苷酸序列如seq id no:53所示。
14.优选地,所述dna聚合酶cgl1289m中rbs的核苷酸序列如seq id no:38所示。
15.优选地,所述cgl1289m和ugi共用一个启动子ptac;所述pmcda1和tada-abe8e共用一个启动子psod。
16.本发明还提供了一种编码上述突变体的核苷酸。
17.优选地,mp1~mp5和mp5t的核苷酸序列如seq id no:55、seq id no:56、seq id no:57、seq id no:58、seq id no:59和seq id no:54所示。
18.本发明还提供了一种含有上述所述的突变体或核苷酸的表达载体。
19.优选地,所述表达载体包括pxmj19、pdxw10、pec-xk99e中的任意一种。
20.本发明还提供了一种重组菌,所述重组菌表达上述的突变体、核苷酸或表达载体。
21.优选地,所述重组菌的出发菌株为谷氨酸棒杆菌13032。
22.本发明还提供了一种谷氨酸棒杆菌高效突变体系,所述突变体系包括上述突变体和诱变剂。
23.本发明还提供了一种提高上述突变体诱变效率的方法,包括如下步骤:所述突变体在诱导剂浓度为0.5~1.5mm,诱导温度为25~35℃下诱导。
24.本发明还提供了一种上述重组菌的构建方法,包括如下步骤:扩增突变体的核苷酸序列,连接到表达载体上,然后将表达载体转化到谷氨酸棒杆菌中,得到重组菌。
25.本发明还提供了一种上述mp5t在制备质粒消除体系中的应用。
26.优选地,所述质粒消除体系还包括诱变剂。
27.优选地,所述mp5t在培养温度为25~35℃条件下实现质粒的正常复制,当培养温度为40~45℃时,质粒无法正常复制,从而实现突变质粒的消除。
28.本发明还提供了一种上述突变体、核苷酸、表达载体或突变体系在筛选高产谷氨酸底盘细胞中的应用。
29.本发明还提供了一种上述突变体、核苷酸、表达载体或突变体系在生产氨基酸中的应用。
30.本发明还提供了一种上述突变体、核苷酸、表达载体或突变体系在筛选耐酸型菌株中的应用。
31.优选地,所述耐酸性菌株为高产谷氨酸耐酸性菌株。
32.相对于现有技术,本发明具有如下有益效果:
33.(1)本发明提供了一种谷氨酸棒杆菌高效突变体,本发明的突变体具备了高效的突变率以及广阔的突变谱,尤其是突变体mp5t还具备了完成突变株筛选后,实现质粒的消除,不会使得突变质粒继续留存在细胞内持续造成本底突变,也为后续菌株的代谢工程改造以及遗传改造奠定了基础。
34.(2)与常规突变方法(如化学诱变、紫外诱变、artp诱变)相比,mp5t比常规诱变方法具有更高的突变率,突变频率f高达1.66
×
10-3
,也就是说,1000个突变子中就能得到1.66个阳性突变子。而且,本发明的突变体的诱变方法简单,而且可以使持续在细胞内实现突变的积累而不对操作者造成任何伤害,极大的缩短突变株文库构建时间,避免了资源和能源的浪费,大大提高了工作效率。
35.(3)本发明利用突变体筛选得到耐酸型突变菌株且在一定程度上提高了l-谷氨酸的发酵水平,相对于正常的实验室进化筛选,缩短了菌种选育的时间,同时也为后续系统代谢工程改造打造谷氨酸高产菌株提供了耐酸型底盘细胞。
附图说明
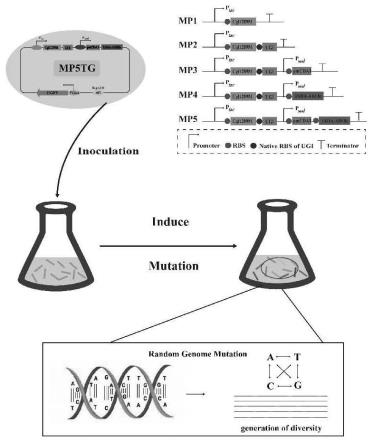
36.图1为谷氨酸棒杆菌高效诱变体系的构建示意图;
37.图2为dna聚合酶dnaq926与cgl1289同源性比对;
38.图3为dna聚合酶cgl1289和cgl1243的exo i保守区域序列比对
39.图4为c.glutamicum 13032耐受利福平抗生素浓度曲线;
40.图5为重组菌cg-mp1、cg-mp-cgl1289的细胞存活情况,其中ck组为不添加iptg的对照组;
41.图6为重组菌cg-mp1、cg-mp-cgl1243m、cg-mp-cgl1289和cg-mp-cgl1243的突变率μ
bp
以及细胞存活率结果图;
42.图7中a为mp1、mp2、mp3、mp4、mp5突变质粒的结构;b为mp1、mp2、mp3、mp4、mp5突变质粒的作用下的细胞存活率;c为c.glutamicum中mp1、mp2、mp3、mp4、mp5的突变率μ
bp
;d为cg-mp1、cg-mp2、cg-mp3、cg-mp4和cg-mp5的细胞存活情况;
43.图8为mps突变质粒突变谱的测定,a为mp1、mp3、mp4和mp5突变谱测定;b为mp1、mp3、mp4和mp5中rpob基因突变频率测定;
44.图9为不同强度启动子对突变质粒mps突变率的影响;
45.图10为不同温度和不同浓度的iptg对突变质粒mp5-rbs6突变率的影响;
46.图11为突变体质粒mp5t的消除,a为不含氯霉素抗性的bhi平板上的突变体质粒mp5t的消除情况;b为含有氯霉素抗性的bhi平板上的突变体质粒mp5t的消除情况;
47.图12为mpt5与其他常规诱变技术的比较,a为不同诱导条件下耐受利福平抗性菌株出现的频率;b~c为artp、ems和mnng诱变产生的突变谱图;
48.图13为耐酸突变株的筛选,a为bhi(ph 5.3)固体平板上单菌落耐酸性生长检测结果;b为耐酸突变株ns-a1、ns-b4、ns-d1和野生型c.glutamicum 13032细胞存活率测定结果;c为耐酸突变株ns-a1、ns-b4、ns-d1和野生型c.glutamicum 13032细胞在bhi平板进行点板试验测得的细胞存活情况;
49.图14为c.glutamicum 13032和耐酸突变株ns-a1的发酵动力学参数。
具体实施方式
50.本发明提供了一种谷氨酸棒杆菌高效突变体,所述突变体包括mp1~mp5和mp5t中的任意一种:
51.所述mp1包括dna聚合酶cgl1289m,所述dna聚合酶cgl1289m的氨基酸序列如seq id no:13所示;
52.所述mp2包括如下元件:所述cgl1289m和尿嘧啶dna糖基化酶抑制剂ugi;所述cgl1289m的下游串联ugi,所述ugi的氨基酸序列如seq id no:25所示;
53.所述mp3包括如下元件:所述cgl1289m、ugi和胞嘧啶脱氨酶pmcda1;所述cgl1289m、ugi和pmcda1依次串联;所述pmcda1的氨基酸序列如seq id no:27所示;
54.所述mp4包括如下元件:所述cgl1289m、ugi和腺嘌呤脱氨酶tada-abe8e;所述cgl1289m、ugi和tada-abe8e依次串联,所述tada-abe8e的氨基酸序列如seq id no:29所示。
55.所述mp5包括如下元件:所述cgl1289m、ugi、pmcda1和tada-abe8e;所述cgl1289m、ugi、pmcda1和tada-abe8e依次串联;
56.所述mp5t包括如下元件:温度敏感型复制起始位点repa101、所述cgl1289m、ugi、pmcda1和tada-abe8e;所述repa101、cgl1289m、ugi、pmcda1和tada-abe8e依次串联,所述repa101的核苷酸序列如seq id no:53所示。
57.在本发明中,通过对c.glutamicum来源的dna聚合酶进行比对分析,发现dna聚合酶cgl1289具有高度保守的exo i区域,且所包含的exo i区域中具有两个高度保守的天冬氨酸和谷氨酸残基,本发明把dna聚合酶cgl1289的exo i区域中的两个高度保守的天冬氨酸和谷氨酸残基都突变为丙氨酸之后,即dna聚合酶cgl1289m,使得细胞的突变率和致死率显著提高。
58.在本发明中,所述dna聚合酶cgl1289m中rbs的核苷酸序列优选地如seq id no:38所示,进一步提高了突变体的突变率。
59.在本发明中,所述cgl1289m和ugi共用一个启动子ptac;所述pmcda1和tada-abe8e共用一个启动子psod,本发明曾尝试利用弱诱导型启动子parac来表达dna聚合酶cgl1289m,但在parac调控下,并不能发挥突变能力,但本发明直接选择强启动子ptac调控dna聚合酶cgl1289m的表达,成功使得突变体发挥了突变功能,同时选择启动子调控psodpmcda1和tada-abe8e表达有利于提高谷氨酸棒杆菌的突变率。
60.在本发明中,所述尿嘧啶dna糖基化酶抑制剂ugi来源于枯草芽孢杆菌噬菌体pbs2,能够在c.glutamicum 13032中通过抑制dna糖基化酶的活性,扰乱碱基切除修复系统并且能够提高突变率以及降低细胞存活率。
61.在本发明中,所述胞嘧啶脱氨酶pmcda1来源于petromyzon marinus的胞嘧啶脱氨酶pmcda1可在c.glutamicum中表达,并能增加碱基对g-c到a-t的突变频率;腺嘌呤脱氨酶tada-abe8e为david liu团队从大肠杆菌中进化而来,能够在c.glutamicum中以dna为底物实现腺嘌呤脱氨基。
62.在本发明中,当碱基切除修复系统即ugi和dna聚合酶丧失校对核酸外切酶活性时,胞嘧啶脱氨酶pmcda1和腺嘌呤脱氨酶tada-abe8e的表达会显著增加dna复制过程中的突变率以及拓宽碱基突变谱。
63.在本发明中,mp5t包括如下元件:温度敏感型复制起始位点repa101、dna聚合酶cgl1289、尿嘧啶dna糖基化酶抑制剂ugi、胞嘧啶脱氨酶pmcda1、腺嘌呤脱氨酶tada-abe8e,各元件之间具有协同增效的作用,可显著提高谷氨酸棒杆菌的突变率。
64.在本发明中,通过对上述突变体的突变谱测定,本发明的上述突变体不仅拥有高的突变率,同时也具备广泛的突变谱。
65.本发明还提供了一种编码上述突变体的核苷酸。
66.在本发明中,mp1~mp5和mp5t的核苷酸序列优选地如seq id no:55、seq id no:56、seq id no:57、seq id no:58、seq id no:59和seq id no:54所示。
67.本发明还提供了一种含有上述所述的突变体或核苷酸的表达载体。
68.在本发明中,所述表达载体优选地包括pxmj19、pdxw10、pec-xk99e中的任意一种,进一步优选地为pxmj19。
69.本发明还提供了一种重组菌,所述重组菌表达上述的突变体、核苷酸或表达载体。
70.在本发明中,所述重组菌的出发菌株优选为c.glutamicum 13032,购自北纳生物。
71.本发明还提供了一种上述重组菌的构建方法,包括如下步骤:扩增突变体的核苷酸序列,连接到表达载体上,然后将表达载体转化到谷氨酸棒杆菌中,得到重组菌。
72.本发明还提供了一种谷氨酸棒杆菌高效突变体系,所述突变体系包括上述突变体和诱变剂。
73.本发明还提供了一种提高上述突变体诱变效率的方法,包括如下步骤:所述突变体在诱导剂浓度为0.5~1.5mm,诱导温度为25~35℃下诱导。
74.在本发明中,当所述突变体为mp5,诱导剂浓度为1.0mm,诱导温度为30℃时,诱变效率达到最高,突变率μbp达到了6.12
×
10-6
,相对于野生型谷氨酸棒杆菌提高了153000倍。
75.本发明还提供了一种上述mp5t在制备质粒消除体系中的应用。
76.在本发明中,所述质粒消除体系还包括诱变剂。在本发明中,所述诱变剂优选为iptg。所述mp5t在培养温度为25~35℃条件下实现质粒的正常复制,当培养温度为40~45℃时,质粒无法正常复制,从而实现突变质粒的消除。在本发明一优选的实施方式中,所述mp5t在培养温度为28~32℃条件下实现质粒的正常复制,当培养温度为41~43℃时,质粒无法正常复制,从而实现突变质粒的消除。
77.本发明还提供了一种上述突变体、核苷酸、表达载体或突变体系在筛选高产谷氨酸底盘细胞中的应用。
78.本发明还提供了一种上述突变体、核苷酸、表达载体或突变体系在生产氨基酸中的应用。
79.在本发明中,所述氨基酸优选地包括谷氨酸、缬氨酸、赖氨酸中的一种或多种。
80.本发明还提供了一种上述突变体、核苷酸、表达载体或突变体系在筛选耐酸型菌株中的应用。
81.在本发明中,所述耐酸性菌株优选为高产谷氨酸耐酸性菌株。在本发明中,本发明的耐酸型菌株能够在ph≤5.3条件下正常生长,且提高了l-谷氨酸的产量。
82.下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
83.下述实施例中涉及的培养基如下:
84.lb培养基(g/l),用于e.coli培养:yeast extract 5,tryptone 10,nacl 10,ph 7.0,121℃灭菌20min,根据需要添加至所需要的抗生素浓度。如需配置固体培养基,则需要加入1.5~2%的琼脂粉。
85.bhi培养基(g/l),用于c.glutamicum细胞培养:bhi固体粉末38.5,ph 7.0,115℃灭菌20min,根据需要添加至所需抗生素浓度。如需配置固体培养基,则需要加入1.5~2%的琼脂粉。
86.bhigs感受态培养基(g/l),用于制备c.glutamicum感受态细胞:bhi固体粉末18.5,甘氨酸30,tween 801,d-山梨醇91,115℃灭菌20min。
87.发酵种子培养基(g/l):葡萄糖25,k2hpo41.5,mgso40.6,玉米浆30,feso4·
7h2o 0.005,mnso4·
h200.005,尿素2.5(单独灭菌),ph 7.3。
88.发酵培养基(g/l):葡萄糖140,k2hpo41,mgso40.6,玉米浆5,feso4·
7h2o 0.005,mnso4·
h200.005,尿素7(单独灭菌),ph 7.3。
89.补料培养基(g/l):葡萄糖800。
90.下述实施例中涉及的突变率及致死率计算方法如下:
91.碱基突变率计算公式:
92.μ
bp
=f/[r
×
ln(n/n0)]。μ
bp
表示每代碱基突变率,f表示利福平耐受突变体频率(f=p/p0,p表示添加iptg诱导后在氯霉素和利福平抗生素双抗平板上的菌落数,p0表示添加iptg诱导后在氯霉素单抗平板上生长的菌落数),r代表产生利福平耐药性特殊位点的突变个数,n表示利福平抗性平板上最终的单克隆个数,n0表示刚观察到单克隆生长时的利福平抗性平板上的单克隆个数。
[0093]
致死率计算公式:
[0094]
μ=m/m0,其中添加iptg诱导的氯霉素平板上的菌落数为m,未添加iptg诱导的氯霉素平板上的菌落数m0。
[0095]
实施例1
[0096]
1.1c.glutamicum内源性dna聚合酶的筛选
[0097]
大肠杆菌体系虽然具备了高效的突变体系mp6,但研究发现,mp6突变质粒在c.glutamicum 13032中基本不能够发挥其高效的诱变能力。影响mp6突变质粒的关键酶是dna聚合酶dnaq926,通过将dna聚合酶dnaq926与c.glutamicum内源性的dna聚合酶进行序列比对分析,发现同源性最高的dna聚合酶为cgl1289,且同源性只有18.1%(见图2)。猜测可能是danq926的低序列同源性,限制了mp6在c.glutamicum 13032中的突变率。因此,寻找c.glutamicum内源性的dna聚合酶,为在c.glutamicum中构建高效突变体系构建基础。
[0098]
通过对c.glutamicum来源的dna聚合酶进行比对分析,发现dna聚合酶cgl1289和cgl1243具有高度保守的exo i区域(所述dna聚合酶cgl1289的氨基酸序列如seq id no:1所示,所述cgl1243的氨基酸序列如seq id no:2所示),且所包含的exo i区域中具有两个高度保守的天冬氨酸和谷氨酸残基(见图3)。
[0099]
本实施例研究表明,当把exo i区域中的两个高度保守的天冬氨酸和谷氨酸残基都突变为丙氨酸之后,会使得细胞的突变率和致死率显著提高。因此,分别将cgl1289和cgl1243两个dna聚合酶的天冬氨酸残基和谷氨酸残基都突变为丙氨酸,具体流程如下:
[0100]
以c.glutamicum 13032基因组为模板,分别以cgl1289-f/r和cgl1243-f/r为引物
pcr扩增出dna聚合酶cgl1289和cgl1243基因片段。以pxmj19质粒为模板,利用引物对p19 f/p19 r反向pcr扩增pxmj19线性化载体。而后将cgl1289和cgl1243基因片段分别与线性化载体pxmj19进行同源重组连接,化转至e.coli bl21感受态。菌落pcr鉴定正确的阳性转化子并送测序公司测序验证,比对测序得到的结果,验证无误的重组载体即为mp-cgl1289和mp-cgl1243。而后将cgl1289和cgl1243两个dna聚合酶的天冬氨酸残基和谷氨酸残基都突变为丙氨酸,再分别利用引物对cgl1289m-f/r和cgl1243m-f/r分别以表达载体mp-cgl1289和mp-cgl1243为模板反向pcr。按相同方法构建获得重组载体mp1和mp-cgl1243m。将重组质粒mp1、mp-cgl1243m、mp-cgl1289和mp-cgl1243电转化转化到c.glutamicum 13032中,构建重组菌cg-mp1、cg-mp-cgl1243m、cg-mp-cgl1289和cg-mp-cgl1243。其中,cgl1289-f/r、cgl1243-f/r、p19 f/p19 r、cgl1289m-f/r和cgl1243m-f/r引物序列具体如下:
[0101]
cgl1289-f:aaacagaattaattaagcttaaaggaggacaactagtgttggggcgtcgaaaag(seq id no:3)
[0102]
cgl1289-r:gagtcgacctgcaggcatgcctattcccaaatatctttgagggtgttcg(seq id no:4)
[0103]
cgl1243-f:aaacagaattaattaagcttaaaggaggacaactaatgaactcaccaagcaatccat(seq id no:5)
[0104]
cgl1243-r:gagtcgacctgcaggcatgcttaggatgcggggtcagtt(seq id no:6)
[0105]
p19-f:gcatgcctgcaggtcgac(seq id no:7)
[0106]
p19-r:aagcttaattaattctgtttcctgtgtga(seq id no:8)
[0107]
cgl1289m-f:ctgtggccgtggcaacgac(seq id no:9)
[0108]
cgl1289m-r:gtcgttgccacggccacag(seq id no:10)
[0109]
cgl1243m-f:ctccttcgccctggcaacaa(seq id no:11)
[0110]
cgl1243m-r:ttgttgccagggcgaaggag(seq id no:12)
[0111]
所述dna聚合酶cgl1289m的氨基酸序列如seq id no:13所示,cgl1289m核苷酸序列如seq id no:14所示;所述dna聚合酶cgl1243m的氨基酸序列如seq id no:15所示,cgl1243m核苷酸序列如seq id no:16所示。
[0112]
基于筛选rifr突变体的rpob/rifr系统已经成功应用于e.coli、c.glutamicum等的突变率的计算。因此,为了测定重组菌cg-mp1、cg-mp-cgl1243m、cg-mp-cgl1289和cg-mp-cgl1243的突变率,首先要测定野生型c.glutamicum 13032耐受利福平抗性的最大抑菌浓度。于是取培养至对数中期的c.glutamicum 13032细胞,并分别在含有0μg/ml、0.5μg/ml、1μg/ml、1.5μg/ml、2μg/ml和4μg/ml的利福平抗性固体平板上划线以及利用灭菌的生理盐水进行10倍稀释点板。
[0113]
计算细胞存活率发现,当添加利福平抗性浓度为2μg/ml时,c.glutamicum 13032的细胞存活率仅为1.35%,当添加利福平抗性浓度为4μg/ml时,c.glutamicum 13032的细胞基本不能存活(见图4)。因此,后续试验测定突变率所选取的利福平浓度为10μg/ml,以保证只有获得了利福平抗性突变菌株方能存活。
[0114]
将野生型c.glutamicum 13032、重组菌cg-mp1、cg-mp-cgl1243m、cg-mp-cgl1289和cg-mp-cgl1243在30℃,180rpm条件下培养,当处于对数中期时,分别添加0.5mm iptg诱导表达,继续在30℃,180rpm条件下培养24~36h,将细胞培养物10倍梯度稀释并涂布到含
有15μg/ml氯霉素和10μg/ml利福平的固体bhi双抗平板和只含有氯霉素抗性的bhi固体平板,并于30℃恒温培养箱静置培养24h。
[0115]
结果表明,重组菌cg-mp1的细胞存活率仅有82.4%,突变率μ
bp
达到了1.2
×
10-7
,相对于野生型对照菌株c.glutamicum 13032提高了3000倍,重组菌cg-cgl1289由于没有将exo i区域中的天冬氨酸残基和谷氨酸残基突变为丙氨酸,所以细胞存活率和突变率基本变化不大,但是重组菌cg-1243m并没有像预期一样具有很高的突变率(见图5和图6)。说明cgl1289的exo i区域中的天冬氨酸残基和谷氨酸残基突变为丙氨酸是显著提高突变率的关键。
[0116]
1.2突变体元器件筛选组装
[0117]
以质粒mp6(购自addgene)为模板,以ugi-f/r为引物,pcr扩增获得尿嘧啶dna糖基化酶抑制剂ugi基因片段。在mp1基础上,在cgl1289m的下游串联表达尿嘧啶dna糖基化酶抑制剂ugi,构建重组载体mp2。分别以质粒mp6和苏州金唯智生物有限公司合成的质粒pet28a-tada-abe8e为模板,pmcda1-f/r和tada-f/r为引物pcr分别扩增获得胞嘧啶脱氨酶pmcda1和腺嘌呤脱氨酶tada-abe8e基因片段,pmcda1-tada f/r扩增获得胞嘧啶脱氨酶pmcda1和腺嘌呤脱氨酶tada-abe8e串联的基因片段。而后在mp2的基础上,在ugi的下游继续串联表达胞嘧啶脱氨酶pmcda1和腺嘌呤脱氨酶tada-abe8e,具体地,分别在ugi的下游继续串联表达胞嘧啶脱氨酶pmcda1、在ugi的下游继续串联腺嘌呤脱氨酶tada-abe8e、在ugi的下游继续串联表达胞嘧啶脱氨酶pmcda1和腺嘌呤脱氨酶tada-abe8e,分别构建重组载体mp3、mp4、mp5。重组质粒mp2、mp3、mp4、mp5分别电转化到c.glutamicum 13032中构建获得重组菌cg-mp2、cg-mp3、cg-mp4和cg-mp5。其中胞嘧啶脱氨酶pmcda1和腺嘌呤脱氨酶tada-abe8e受到启动子p
sod
所调控。其中,ugi-f/r、pmcda1-f/r、tada-f/r和pmcda1-tada f/r引物序列具体如下:
[0118]
ugi-f:aaaattaggaggaatttcaacatgacaaatttatc(seq id no:17)
[0119]
ugi-r:gctcggtacccggggatcctttataacattttaattttattttctccattactgtct(seq id no:18)
[0120]
pmcda1-f:tacgaaaggattttttacccaaaggaggacaactaatgaccgacgcggaatacg(seq id no:19)
[0121]
pmcda1-r:gctcggtacccggggatcctttaaaccgccggagatttg(seq id no:20)
[0122]
tada-f:tacgaaaggattttttacccaaaggaggacaactaatgtctgaagtggagttctccc(seq id no:21)
[0123]
tada-r:gctcggtacccggggatcctttaattgatggaggactgtgcct(seq id no:22)
[0124]
pmcda1-tadaf:ccaaatctccggcggtttaaaaaggaggacaactaatgtctgaagtggagttctcc c(seq id no:23)
[0125]
pmcda1-tada r:gctcggtacccggggatcctttaattgatggaggactgtgcct(seq id no:24)
[0126]
所述尿嘧啶dna糖基化酶抑制剂ugi的氨基酸序列如seq id no:25所示,所述ugi的核苷酸序列如seq id no:26所示;所述胞嘧啶脱氨酶pmcda1的氨基酸序列如seq id no:27所示,所述pmcda1的核苷酸序列如seq id no:28所示;所述腺嘌呤脱氨酶tada-abe8e的氨基酸序列如seq id no:29所示,所述tada-abe8e的核苷酸序列如seq id no:30所示。
[0127]
所述mp1的核苷酸序列如seq id no:55所示;所述mp2的核苷酸序列如seq id no:56所示;所述mp3的核苷酸序列如seq id no:57所示;所述mp4的核苷酸序列如seq id no:58所示;所述mp5的核苷酸序列如seq id no:59所示。
[0128]
结果表明,mp2的细胞存活率仅仅只有35.7%,突变率μ
bp
为3.6
×
10-7
,相对于mp1提高了3倍;mp3的细胞存活率为2.4%,突变率μ
bp
达到了2.76
×
10-6
,相对于mp1提高了23倍;mp4的细胞存活率为3%,突变率μ
bp
为1.8
×
10-6
,相对于mp1提高了15倍;mp5的细胞存活率仅仅只有1.7%,突变率μ
bp
达到了3.84
×
10-6
,相对于mp1提高了32倍。总体而言,在cgl1289m和ugi的协同作用下,pmcda1和tada-abe8e可显著提高c.glutamicum的突变率(见图7)。
[0129]
1.3突变质粒突变谱测定
[0130]
利用突变后利福平抗性平板上筛选得到的耐受利福平抗性的突变菌株的利福平抗性编码基因rpob进行高通量测序,分析突变谱。
[0131]
结果显示,发现mp1的突变谱较窄,主要的碱基突变类型集中在t:a-a:t和t:a-c:g。相比之下,mp3的突变碱基分布更均匀,覆盖了更多类型的突变,g:c-a:t的突变显著增加,这是因为dna糖基化酶抑制剂ugi的表达,扰乱了碱基切除修复系统,同时胞嘧啶脱氨酶pmcda1的表达增加了g:c-a:t的突变。同样,mp4由于表达了腺嘌呤脱氨酶tada-abe8e,也具备了比mp1更均匀的碱基突变分布,t:a-c:g和a:t-g:c的突变明显增加。mp5同时表达了胞嘧啶脱氨酶pmcda1和腺嘌呤脱氨酶tada-abe8e,被赋予了更广泛的碱基突变分布,相对于mp1的碱基突变类型,g:c-a:t、t:a-c:g和a:t-g:c的碱基突变类型明显增加(见图8)。
[0132]
同时,基于测序发现的rpob突变位点基本囊括在已经报道了的能够获得利福平抗性的突变位点,其中也发现了三个新突变位点,如g1241a、c1286g和c1313t也能够使得c.glutamicum获得利福平抗性(表1)。这些结果表明,本发明开发的mps突变质粒不仅拥有高的突变率,同时也具备广泛的突变谱。
[0133]
表1利福平耐受突变株rpob基因突变类型
[0134][0135][0136]
注:下划线标注的突变位点表示的是无义突变,加粗字体标注的突变位点表示不包含在已经报道了rpob突变文库中,其与突变位点均为已经报道的能够获得rifr抗性的突变。
[0137]
1.4mp5突变质粒启动子优化
[0138]
前期研究过程中,曾尝试利用弱诱导型启动子p
arac
来表达dna聚合酶cgl1289m,但在p
arac
调控下,并不能发挥突变能力。由于c.glutamicum中蛋白表达量较低,因此直接选择强启动子p
tac
调控dna聚合酶cgl1289m的表达,成功使得突变质粒mp1发挥了突变功能。当dna聚合酶cgl1289m和dna糖基化酶抑制剂ugi处于表达过程中,才会发挥突变能力。因此,这两个酶的表达必须受到严格调控,且在c.glutamicum中能够使用的强诱导型且受到严格调控的启动子稀少,因此,并未对这两个酶的启动子进行优化。
[0139]
为了找到合适的启动子来驱动pmcda1和tada-abe8e的表达,选择了强诱导型启动子(p
tac
)、强组成型启动子(p
tuf
)、中等强度组成型启动子(p
sod
)和弱组成型启动子启动子(p
zwf
)来驱动胞嘧啶脱氨酶pmcda1和腺嘌呤脱氨酶tada-abe8e的表达。
[0140]
以质粒mp5为模板,以p5 f/p5 r为引物,通过反向pcr技术线性化缺失启动子p
sod
基因片段的重组质粒mp5,而后以c.glutamicum 13032基因组为模板分别扩增出启动子p
tuf
和p
zwf
基因片段,而后将启动子p
tuf
和p
zwf
基因片段和线性化mp5质粒进行同源重组连接,并转化e.coli bl21感受态细胞,分别构建重组质粒mptuf和mpzwf。重组质粒mptac以及mpdtac的构建直接以重组质粒mp5为模板,设计相应的引物反向pcr扩增得到相应的线性化片段,而后利用同源重组连接试剂盒将所得到的线性化片段在37℃条件下孵育30min使其自连,而后化转到e.coli bl21感受态细胞,分别构建重组质粒mptac和mpdtac。其中,p5 f/p5 r的引物序列具体如下:
[0141]
p5-f:ttataacattttaattttattttctccattactgtct(seq id no:31)
[0142]
p5-r:aaaggaggacaactaatgaccgac(seq id no:32)
[0143]
通过将构建成功的重组质粒电转到c.glutamicum中并测定突变率发现,无论是强启动子p
tac
、p
tuf
,还是弱启动子p
zwf
,其突变率相对于mp5都没有明显提高。也就是说,适度强度的pmcda1和tada-abe8e表达将有利于提高谷氨酸棒杆菌的突变率(见图9)。
[0144]
1.5rbs序列优化提高mp5突变率
[0145]
利用在线网站rbs calculator(https://www.denovodna.com/software/pre dict_rbs_calculator)针对dna聚合酶cgl1289m设计不同翻译起始率的rbs序列(表2),以替换重组质粒mp5上cgl1289m原始的rbs序列,所用反向pcr引物见表3,以mp5质粒为模板,利用相应的反向pcr引物进行pcr扩增,获得线性化片段并自连,分别构建出重组质粒mp5-rbs1、mp5-rbs2、mp5-rbs3、mp5-rbs4、mp5-rbs5和mp5-rbs6。测序验证正确后,将重组质粒分别电转至c.glutamicum感受态细胞,构建重组菌cg-mp5-rbs1、cg-mp5-rbs2、cg-mp5-rbs3、cg-mp5-rbs4、cg-mp5-rbs5和cg-mp5-rbs6。
[0146]
表2不同翻译起始速率的rbs序列
[0147][0148]
表3不同rbs序列所用反向pcr引物
[0149][0150]
表4不同强度rbs条件下的突变率和细胞存活率
[0151][0152]
测定突变率发现,突变质粒mp5-rbs6的突变率最高,达到5.76
×
10-6
,相对于mp5提高了1.5倍(表4)。
[0153]
1.5mp5-rbs6突变质粒诱导条件优化
[0154]
mps突变质粒需要添加诱导剂iptg来诱导dna聚合酶cgl1289m和dna糖基化酶抑制剂ugi的表达,因而mps的突变率会受到诱导剂的浓度和诱导温度的影响。因此,考察了不同浓度的iptg添加量和诱导温度对于mp5-rbs6突变质粒突变率的影响。
[0155]
mp5-rbs6突变质粒诱导方法:将含mp5-rbs6突变质粒的c.glutamicum在含氯霉素抗性的平板上划线活化,30℃培养箱倒置培养24~48h,挑取单菌落接种添加氯霉素的10ml bhi液体培养基,30℃、180rpm培养24h,按1%接种量转接添加氯霉素抗性的50ml bhi液体培养基,30℃、180rpm继续培养3h后添加不同量iptg,放置不同温度下诱导12h。
[0156]
考察不同诱导温度(16、20、25、30、35℃)对诱变效率的影响时,iptg的添加浓度均控制在0.5mm。
[0157]
结果表明,当诱导温度最适温度为30℃,诱变效率最高,也就是c.glutamicum最适生长的温度(图10中的a和c)。可能是因为无论是高温或者是低温都不利于c.glutamicum生长,同样也会阻碍dna的正常复制,所以会导致突变率降低。
[0158]
考察iptg添加浓度(0.1、0.5、1.0、1.5、2.0mm)对诱变效率的影响时,诱导温度均控制在30℃。
[0159]
结果显示,当iptg的添加浓度为1mm时,诱变效率最高,突变率μ
bp
达到了6.12
×
10-6
(图10中的b和d)。
[0160]
综上,当诱导剂添加浓度为1mm、诱导温度30℃时,mp5-rbs6质粒的诱变效率达到最高,突变率μ
bp
达到了6.12
×
10-6
,相对于野生型谷氨酸棒杆菌提高了153000倍。
[0161]
1.7突变质粒消除体系构建
[0162]
利用crispr体系中的温度敏感型复制起始位点repa101替换mp5-rbs6自身的原始复制起始位点,构建重组菌cg-mp5t。具体构建流程如下:
[0163]
以质粒pcas9为模板,以repa101-f/repa101-r为引物pcr扩增出repa 101基因片段,胶回收纯化获得的repa 101基因片段与反向pcr扩增出的缺失原始质粒复制起始位点的线性化mp5片段进校同源重组连接,连接产物转化至e.colibl21感受态细胞,构建重组质粒mp5t。测序验证正确后,将重组质粒mp5t电转至c.glutamicum感受态细胞,构建重组菌cg-mp5t。其中,
[0164]
repa101-f:tcagatccttccgtatttagccag(seq id no:51)
[0165]
repa101-r:atgtctgaattagttgttttcaaagcaaat(seq id no:52)
[0166]
所述温度敏感型复制起始位点repa101的核苷酸序列如seq id no:53所示;所述mp5t的核苷酸序列如seq id no:54所示。
[0167]
重组菌cg-mp5t在bhi固体平板上划线活化,挑取单菌落转接到含有氯霉素抗性的bhi培养基中,30℃,180rpm培养18~24h。而后转接到无抗bhi培养基中,于42℃、180rpm条件下继续培养12h后,在无抗bhi平板上划线,30℃恒温培养箱中继续培养24~36h。最后,将生长在无抗bhi平板上的单菌落分别一一对应点在无抗bhi培养平板和含有氯霉素bhi培养平板上,30℃培养箱继续培养18~24h。其中,对应编号中氯霉素平板上不生长,无抗平板上生长的菌株即为mp5t质粒成功丢失的菌株。
[0168]
实验表明,mp5t在30℃条件下可正常复制,42℃条件下则很容易就能够实现质粒消除(见图11)。也就是说,mp5t的成功构建使得mp5突变质粒不仅具备了高效的突变率以及广阔的突变谱,同时也具备了完成突变株筛选后,实现质粒的消除,不会使得突变质粒继续留存在细胞内持续造成本底突变,也为后续菌株的代谢工程改造以及遗传改造奠定了基础。
[0169]
1.8mp5t突变质粒与常规突变方法的比较
[0170]
传统的诱变方式主要包括物理和化学诱变方法,为了更直观的比较mp5t与常规诱变方法的突变能力,进一步分析了ems、mnng、uv和artp四种诱变方法在c.glutamicum中的突变效果。
[0171]
(1)甲基磺酸乙酯(ems)诱变:
[0172]
取1ml生长至对数中期c.glutamicum细胞离心并弃除上清液,并用1ml 100mm tris-hcl(ph 7.0)缓冲液在冰上洗涤2次,然后添加100μl ems并用移液枪轻轻吹吸混合均
匀,并在200rpm,30℃条件下处理45min。而后离心并利用1ml 100mm tris-hcl(ph 7.0)缓冲液在冰上洗涤2次以去除ems残留,并将细胞浓度稀释10倍。而后接种于新鲜灭菌且不添加抗生素的bhi培养基中于180rpm,30℃条件下培养12-16h。
[0173]
(2)1-甲基-3-硝基-1-亚硝基胍(mnng)诱变:
[0174]
取1ml生长至对数中期c.glutamicum细胞离心并弃除上清液,并用1ml 200mm乙酸钠(ph 5.5)在冰上洗涤2次,然后添加至终浓度为1.6μg/ml mnng并用移液枪轻轻吹吸混合均匀,并在30℃水浴条件下处理30min。而后收集细胞离心弃除mnng上清,并利用1ml 100mm磷酸钾缓冲液(ph 7.0)在冰上洗涤2次以去除mnng残留,并将细胞浓度稀释10倍。而后接种于新鲜灭菌且不添加抗生素的bhi培养基中于180rpm,30℃条件下培养12~16h。
[0175]
(3)紫外(uv)诱变:
[0176]
取1ml生长至对数中期c.glutamicum细胞离心并弃除上清液,并用1ml 100mm磷酸钾缓冲液(ph 7.0)在冰上洗涤2次,并利用1ml 100mm mgso4溶液重悬细胞。而后将细胞放置于灭菌的空培养皿中并在紫外线条件下处理100s(紫外灯距离培养皿高度为10cm)。处理结束后立刻转移细胞至新鲜灭菌且不添加抗生素的bhi培养基中于180rpm,30℃条件下培养12~16h,培养过程中要用锡箔纸包裹,防止见光发生回复突变。
[0177]
(4)artp诱变:
[0178]
取1ml生长至对数中期c.glutamicum细胞离心并弃除上清液,并用1ml无菌生理盐水在冰上洗涤2次。然后用移液枪吸取10μl细胞悬液并注入到灭菌的小铁片表面,并在artp作用30s。然后取出带有细胞悬液的小铁片并转移至新鲜灭菌且不添加抗生素的bhi培养基中于180rpm,30℃条件下恢复培养2~3h。
[0179]
(5)mp5t诱变:
[0180]
将含有mp5t质粒的c.glutamicum 13032细胞生长至对数中期,添加0.5mm iptg诱导mp5t质粒表达,继续在30℃,180rpm条件下培养24~36h。
[0181]
上述诱变方法所得到的细胞,经过细胞培养之后分别经过10倍梯度稀释涂布于无抗bhi固体平板以及含有终浓度10μg/ml利福平抗生素抗性平板上,倒置于30℃恒温培养箱静置培养24~36h之后,计算每个利福平抗性bhi平板和无抗bhi平板上生长的菌落数,并计算突变率。
[0182]
结果表明,mp5t比其他四种诱变方法具有更高的突变率,突变频率f高达1.66
×
10-3
,也就是说,1000个突变子中就能得到1.66个阳性突变子。化学诱变剂ems和mnng在c.glutamicum中的突变频率不是很高,也有相关文献报道其突变谱比较窄,突变类型并不丰富(见图12)。而且,化学诱变剂如ems和mnng都是强致癌物,在环境中难以降解,对环境和人体具有持续的毒性作用,有着一定的潜在危险。虽然artp诱变具有相对广泛的突变谱(见图12),但其突变率与化学诱变剂ems和mnng的差异不大,在c.glutamicum的突变率仍然较低。在利用紫外诱变筛选利福平耐受突变株的过程中,我们基本上没有得到阳性突变株,这可能是由于紫外诱变效率较低且突变方向具有序列偏好性,主要集中在c:g-a:t的突变。虽然与化学诱变剂相比,artp和uv诱变相对安全可靠,但uv和artp诱变通常构成单轮诱变,不能像化学诱变剂一样可以实现连续诱变。相比之下,mp5t突变质粒不仅突变率高、突变谱广、操作方法简单,而且可以可以使持续在细胞内实现突变的积累而不对操作者造成任何伤害,极大的缩短突变株文库构建时间,避免了资源和能源的浪费,大大提高了工作效率。
[0183]
1.9筛选高产l-谷氨酸底盘细胞
[0184]
基于实验室前期研究,野生型c.glutaminum 13032在初始发酵ph中性时,且发酵过程中不调节ph,当细胞大量合成l-谷氨酸时,细胞微环境中的ph会持续降低,当ph为5.5时,会严重抑制细胞的生长。酸胁迫通常会引起细胞的一系列生理损伤,破坏生物大分子的结构,影响主要代谢途径中酶的活性,导致细胞内物质/能量代谢紊乱。因此,筛选能够耐受更低ph环境的突变菌株,提高细胞的耐酸性,对于c.glutaminum发酵生产l-谷氨酸有着重要的意义。
[0185]
利用mp5t质粒连续诱变处理野生型c.glutaminum 1303236h后,稀释涂布到ph 5.3bhi固体平板上培养3天,而后在平板上挑取了47个单克隆于装有ph 5.3的液体bhi的48深孔板中继续培养24h,然后通过测定细胞密度od
600
。
[0186]
结果表明,突变株ns-a1、ns-b4和ns-d1在ph 5.3环境下的生长状况最佳,生长强度明显优于出发菌株(图13中a)。
[0187]
突变株ns-a1、ns-b4和ns-d1在30℃,180rpm培养12h后,用无菌生理盐水洗涤细胞两次,重悬于带有100mm homo-pipes的bhi(ph 4.0)的培养基中使其初始细胞密度od
600
为0.2,并于30℃,180rpm培养,并分别在培养0h、2h、4h、6h和8h时取样。每个时间所取的细胞样品通过10倍梯度稀释并点板于bhi固体平板上,于30℃恒温培养箱中培养24h。培养结束后取出平板,并计算菌落数以及细胞存活率。细胞存活率=不同时间酸处理细胞数/未进行酸处理的细胞数。
[0188]
结果表明,ns-a1在ph4的条件下胁迫8h,细胞存活率仍然高达43.75%,出发菌株c.glutaminum 13032的细胞存活率仅仅只有0.71%(图13中b)。
[0189]
之后对ns-a1菌株进行5l发酵罐发酵,具体操作如下:取ns-a1保藏菌株冻管,在bhi固体培养皿上划线,分离单菌落。然后在bhi液体培养基中用接种环挑取单菌落并接入其中,于30℃,180rpm条件下培养12h,活化细胞。而后在10ml发酵种子培养基中接入活化后的细胞(接种量1%),并于30℃,180rpm条件下培养12h,然后转移至200ml种子培养基中继续培养16-18h。最后在装有1.8l发酵培养基的5l发酵罐中将培养完成的200ml发酵种子液接全部接入。发酵罐发酵初始条件:30℃,搅拌速率600r/min,通气量1vvm,ph 7.3。当发酵培养基中葡萄糖浓度至20g/l时,发酵过程中的葡萄糖浓度控制在20~30g/l(流加补料培养基控制)。
[0190]
结果显示,耐酸突变株ns-a1发酵48h能够积累l-谷氨酸35.3g/l,相对于野生型c.glutaminum 13032提高了16.7%(见图14)。
[0191]
因此,我们成功利用mp5t,筛选得到了3株能够在ph 5.3条件下正常生长的突变菌株,且在一定程度上提高了l-谷氨酸的发酵水平,相对于正常的实验室进化筛选,缩短了菌种选育的时间,同时也为后续系统代谢工程改造打造l-谷氨酸高产菌株提供了耐酸型底盘细胞。这一谷氨酸棒杆菌高效诱变体系也可应用于其他氨基酸生产中。
[0192]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1