L-薄荷胺、D-新薄荷胺及其制备方法
l-薄荷胺、d-新薄荷胺及其制备方法
技术领域
1.本发明涉及薄荷醇合成制备技术领域,特别是l-薄荷胺、d-新薄荷胺及其制备方法。
背景技术:
2.薄荷醇(又名薄荷脑),是一种具有清凉性的化合物,不同构型的清凉效果不一致,其中又以l-薄荷醇最佳,广泛运用于食品,烟草,医药,日化等行业。但是l-薄荷醇的缺点也比较明显,难溶于水,挥发性强,薄荷味比较重,影响了其进一步的使用空间。为了克服传统薄荷醇的局限性,以薄荷醇的基本骨架衍生的化合物成为新型清凉剂的研发方向,例如薄荷酯,薄荷醚,薄荷酰胺等,其中以手性薄荷胺为分子结构基础的胺基衍生物目前研究较少,具有比较大的发展空间。
3.此外,手性薄荷胺由于其具有三个手性中心的六元环结构,作为手性诱导源,在催化不对称合成中也有着广泛的用途,例如王世海以手性薄荷胺为原料制备的n-薄荷基α-溴代苯乙酰胺,用作非手性单体甲基丙烯酸-1-苯基二苯并环庚醇酯的原子转移自由基聚合的手性引发剂。周宜荣选择手性薄荷胺与各类氨基酸缩合,得到了一系列结构新颖的手性二仲胺配体,用来催化不对称henry反应和michael加成反应。
4.目前手性薄荷胺的合成途径主要是通过酮肟转化,将薄荷醇氧化成薄荷酮,之后与盐酸羟胺反应得到薄荷酮肟,kozlov使用铂黑-冰醋酸体系和雷尼镍-甲醇体系将薄荷酮肟转化为手性薄荷胺,但是产物复杂,非对映体产率接近60%,而schopohl等采用大当量的金属钠(》20倍)在无水乙醇中还原薄荷酮肟,demidova等以au/al2o3为载体,用金纳米颗粒催化氢气还原薄荷酮肟,均可以较高选择性的获得(1r,2r,5r)-手性薄荷胺([α]
d20
=-35.7
°
)。上述路线中使用到了铬试剂和大量的过渡金属催化反应,污染性和实验危险较大,此外也仅能获得一种构型的手性薄荷胺,存在着一定的局限性。
技术实现要素:
[0005]
为了解决现有技术中手性薄荷胺的合成途径存在的污染性和实验危险较大,产物构型单一的问题,本发明的目的之一是提供一种l-薄荷胺的制备方法。
[0006]
为实现上述目的,本发明采用了以下技术方案:一种l-薄荷胺的制备方法,包括如下步骤:
[0007]
步骤1、取l-薄荷醇、对硝基苯甲酸和三苯基膦于反应瓶中,加入四氢呋喃至溶解,冰浴条件下缓慢滴加偶氮二甲酸二乙酯,滴加完毕转移至室温反应8-12h,其中l-薄荷醇与对硝基苯甲酸、三苯基膦、偶氮二甲酸二乙酯的物质的量的比为1:1:1.5:1.5,反应产物经洗涤、萃取、浓缩和纯化,得到式(1a)的d-新薄荷醇酯;
[0008]
步骤2、取式(1a)的d-新薄荷醇酯溶于甲醇中,分批加入碳酸钾,其中d-新薄荷醇酯与碳酸钾的物质的量的比为1:2,室温搅拌下反应2-4h,反应液经过滤、洗涤、萃取和纯化得到式(2a)的d-新薄荷醇;
[0009]
步骤3、取式(2a)的d-新薄荷醇、邻苯二甲酰亚胺、三苯基膦加入到四氢呋喃中溶解,冰浴条件下缓慢滴加偶氮二甲酸二乙酯,其中,d-新薄荷醇、邻苯二甲酰亚胺、三苯基膦和偶氮二甲酸二乙酯的物质的量的比为1:1:1.5:1.5,滴加完毕转移至室温反应8-12h,反应液经洗涤、萃取、浓缩和柱层析纯化,得到式(3a)的l-薄荷酰亚胺中间体;
[0010]
步骤4、取式(3a)中的l-薄荷酰亚胺中间体,加入甲醇至溶解,室温下加入80wt%水合肼溶液,l-薄荷酰亚胺中间体和水合肼溶液中水合肼的物质的量的比为1:5,升温至50-60℃反应4-6h至大量白色固体析出,过滤后调节滤液的ph为酸性,用乙酸乙酯稀释后加水萃取,调节水层的ph为碱性,再用乙酸乙酯分批萃取水层,合并有机层旋干得到无色液体即为式(4a)中的l-薄荷胺;
[0011][0012]
本发明的目的之二是提供一种由上述制备方法制得的l-薄荷胺。
[0013]
本发明的目的之三是提供一种d-新薄荷胺的制备方法。
[0014]
为实现上述目的,本发明采用了以下技术方案:一种d-新薄荷胺的制备方法,包括如下步骤:
[0015]
步骤1、取l-薄荷醇、邻苯二甲酰亚胺、三苯基膦加入到四氢呋喃中溶解,冰浴条件下缓慢滴加偶氮二甲酸二乙酯,其中,l-薄荷醇、邻苯二甲酰亚胺、三苯基膦和偶氮二甲酸二乙酯的物质的量的比为1:1:1.5:1.5,滴加完毕转移至室温反应8-12h,反应液经洗涤、萃取、浓缩和柱层析纯化,得到式(3c)的d-新薄荷酰亚胺中间体;
[0016]
步骤2、取式(3c)中的d-新薄荷酰亚胺中间体,加入甲醇溶解,室温下加入80wt%水合肼溶液,d-新薄荷酰亚胺中间体和水合肼溶液中水合肼的物质的量的比为1:5,升温至50-60℃反应4-6h至大量白色固体析出,过滤后调节滤液的ph为酸性,用乙酸乙酯稀释后加水萃取,调节水层的ph为碱性,再用乙酸乙酯分批萃取水层,合并有机层旋干得到无色液体即为式(4c)中的d-新薄荷胺;
[0017][0018]
本发明的目的之四是提供一种由上述制备方法制得的d-新薄荷胺。
[0019]
本发明的目的之五是提供一种d-新薄荷胺的制备方法。
[0020]
为实现上述目的,本发明采用了以下技术方案:d-新薄荷胺的制备方法,包括如下步骤:
[0021]
步骤1:取l-薄荷醇、三苯基膦加入到四氢呋喃中溶解,冰浴条件下缓慢滴加偶氮二甲酸二乙酯,反应半小时后加入叠氮磷酸二苯酯,并转移至室温反应8-12h,其中,l-薄荷
醇、叠氮磷酸二苯酯、三苯基膦和偶氮二甲酸二乙酯的物质的量的比为1:1.5:3:3,反应液经洗涤、萃取、浓缩和柱层析纯化,得到式(3e)的d-新薄荷叠氮中间体;
[0022]
步骤2、取式(3e)的d-新薄荷叠氮中间体和15wt%的钯碳加入反应瓶中,加入甲醇溶解,在1-2个大气压的氢气下反应24h,混合物用硅藻土过滤并用甲醇洗涤,浓缩后得到无色液体即为式(4c)中的d-新薄荷胺;
[0023][0024]
本发明的目的之六是提供一种由上述制备方法制得的d-新薄荷胺。
[0025]
本发明相比现有技术的有益效果在于:
[0026]
1)传统手性薄荷胺的合成路线如下:
[0027][0028]
通过酮肟转化,将薄荷醇氧化成薄荷酮,之后与盐酸羟胺反应得到薄荷酮肟,再使用薄荷酮肟制备手性薄荷胺。现有技术在使用薄荷酮肟制备手性薄荷胺的过程中均使用铬试剂和大量的过渡金属催化反应,污染性和实验危险较大,此外也仅能获得一种构型的手性薄荷胺,存在着一定的局限性。
[0029]
mitsunobu反应(光延反应)是一种双分子亲核取代反应(sn2反应),在三苯基膦和偶氮二甲酸二乙酯的催化下,仲醇羟基被不同的亲核试剂取代,同时跟羟基所连的碳原子构型发生翻转。本文基于分子结构设计原理,以市场上容易获得的l薄荷醇为原料,与对硝基苯甲酸通过mitsunobu反应生成构型翻转的醇酯中间体,水解得到d-新薄荷醇,之后连同l薄荷醇一起,再次采用mitsunobu反应与邻苯二甲酰亚胺结合得到构型翻转的薄荷酰亚胺中间体,最后肼解得到l-薄荷胺和d-新薄荷胺,或者以l-薄荷醇为原料,与叠氮磷酸二苯酯通过mitsunobu反应生成构型翻转的叠氮中间体,之后钯碳还原得到d-新薄荷胺。
[0030]
本发明制备两种手性薄荷胺的方法具有方法新颖,实验操作简单,对环境友好的优点。以一到两次mitsunobu反应,巧妙的实现了羟基到胺基的官能团转化,并且由于mitsunobu反应构型翻转的特点,获得了确定构型的手性薄荷胺。相比于传统只能获得一种构型手性薄荷胺的方法,本发明一次性制备了两种确定构型的手性薄荷胺,扩充了手性薄荷胺的适用范围;反应条件温和,所有反应温度均不超过60℃,无需氮气保护,与传统方法相比,避免使用了铬和金属钠等污染性大,实验危险性高的试剂。
[0031]
2)本发明制得的两种手性薄荷胺具有应用范围广的优点。作为初始工具分子,可以通过酰胺缩合,还原胺化,亲核取代等方式合成多种不同构型的手性胺基衍生物,可用于后续清凉剂的研发和不对称反应的催化。
附图说明
[0032]
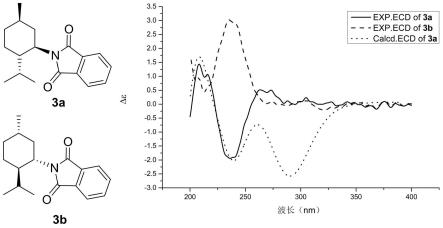
图1是式3a的化合物的计算ecd光谱、实施例1中制得的式3a的实验ecd光谱图以及3a的对映异构体3b的实验ecd光谱图。
具体实施方式
[0033]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明,基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0034]
主要仪器与试剂
[0035]
avance iii型400mh核磁共振波谱仪(瑞士bruker公司);bc-r202b型旋转蒸发器(上海贝凯生物化工设备有限公司);1260/6460液相色谱串联三重四级杆质谱(安捷伦科技有限公司);jasco p-2000旋光仪(日本jasco公司)
[0036]
l-薄荷醇,对硝基苯甲酸,邻苯二甲酰亚胺,三苯基膦,偶氮二甲酸二乙酯,叠氮磷酸二苯酯,80wt%水合肼,四氢呋喃,乙酸乙酯,甲醇,丙酮,氯化钠,碳酸钾,无水硫酸镁(药品均购自麦克林,阿拉丁等品牌试剂公司)。
[0037]
实施例1
[0038]
本实施例提供一种l-薄荷胺的制备方法,包括如下步骤:
[0039]
步骤1,1a的合成。称取10.0g(0.064mol)l-薄荷醇,10.7g(0.064mol)对硝基苯甲酸和25.1g(0.096mol)三苯基膦于250ml反应瓶中,加入40ml四氢呋喃中溶解,冰浴搅拌下缓慢滴加16.8ml(0.096mol)偶氮二甲酸二乙酯,滴加完毕后转移至室温反应12h,tlc监测反应完全。
[0040]
待反应完全将反应液用饱和碳酸氢钠水溶液洗涤,二氯甲烷萃取,浓缩得半固体。将半固体悬浮在100ml乙醚中,加入40ml正己烷,搅拌半小时后抽滤,滤液浓缩得到黄色油状物,即式1a的d-新薄荷醇酯;
[0041]
步骤2,2a的合成。称取8.0g(0.026mol)式1a的d-新薄荷醇酯溶于20ml甲醇中,分批加入7.2g(0.052mol)碳酸钾,室温搅下2h,tlc监测反应完全。将反应液过滤,加水洗涤,二氯甲烷萃取,浓缩后通过硅胶柱层析(pe:ea=5:1,v/v)纯化得到式2a的d-新薄荷醇;
[0042]
步骤3,3a的合成。称取2.5g(0.016mol)式2a的d-新薄荷醇,2.4g(0.016mol)邻苯二甲酰亚胺和6.3g(0.024mol)三苯基膦于100ml反应瓶中,加入10ml四氢呋喃溶解,冰浴搅拌下缓慢滴加4.2ml(0.024mol)偶氮二甲酸二乙酯,滴加完毕后转移至室温反应12h,tlc监测反应完全。反应液经浓缩得粗品,通过硅胶柱层析(pe:ea=20:1,v/v)纯化得到式3a的l-薄荷酰亚胺中间体;
[0043]
步骤4,4a的合成。称取1.0g(0.0035mol)l-薄荷酰亚胺中间体于25ml反应瓶中,加入5ml甲醇溶解,室温下加入0.3ml 80wt%水合肼溶液,之后升温至50℃反应6h至大量白色固体析出,过滤后往滤液中加入稀盐酸调ph为酸性,乙酸乙酯稀释后加水萃取,往水层加入氢氧化钠溶液调ph为碱性,再用乙酸乙酯分批萃取水层,合并有机层旋干得到无色液体即为式4a的l-薄荷胺。
[0044]
(4a)l-薄荷胺
[0045]
无色油状液体。1h nmr(400mhz,cdcl3)δ2.52-2.41(m,1h),2.15-2.00(m,1h),
1.80-1.71(m,1h),1.67-1.52(m,2h),1.43-1.30(m,1h),1.10(s,2h),0.87(d,j=7.2hz,3h),0.84(d,j=6.8hz,3h),0.96-0.70(m,4h),0.72(d,j=7.2hz,3h);
13
c nmr(100mhz,cdcl3)δ51.7,50.7,46.1,35.0,32.2,26.1,23.4,22.5,21.3,15.6.esi-ms:156[m+h]+,[α]d
20
=-33.2
°
(c=1.4in chcl3)
[0046]
经计算,上述实施例的产率为38.6%,手性薄荷胺的纯度为95%。
[0047]
实施例2
[0048]
本实施例提供一种d-新薄荷胺的制备方法,包括如下步骤:
[0049]
步骤1,3c的合成。称取2.5g(0.016mol)l-型薄荷醇,2.4g(0.016mol)邻苯二甲酰亚胺和6.3g(0.024mol)三苯基膦于100ml反应瓶中,加入10ml四氢呋喃溶解,冰浴搅拌下缓慢滴加4.2ml(0.024mol)偶氮二甲酸二乙酯,滴加完毕后转移至室温反应12h,tlc监测反应完全。反应液经浓缩得粗品,通过硅胶柱层析(pe:ea=20:1,v/v)纯化得到式3c的d-新薄荷酰亚胺中间体;
[0050]
步骤2,4c的合成。称取1.0g(0.0035mol)式3c的d-新薄荷酰亚胺中间体于25ml反应瓶中,加入5ml甲醇溶解,室温下加入0.3ml 80wt%水合肼溶液,之后升温至50℃反应6h至大量白色固体析出,过滤后往滤液中加入稀盐酸调ph为酸性,乙酸乙酯稀释后加水萃取,往水层加入氢氧化钠溶液调ph为碱性,再用乙酸乙酯分批萃取水层,合并有机层旋干得到无色液体即为式4c的d-新薄荷胺。
[0051]
(4c)d-新薄荷胺
[0052]
无色油状液体1h nmr(400mhz,cdcl3)δ=2.85-2.83(m,1h),1.68-1.53(m,4h),1.39-1.30(m,1h),1.15-1.02(m,2h),0.95(s,2h),0.87(d,j=6.4hz,3h),0.85(d,j=6.4hz,3h),0.84-0.80(m,1h),0.78(d,j=6.4hz,3h),0.80-0.75(m,1h);
13
c nmr(100mhz,cdcl3)δ48.1,47.5,43.5,35.5,29.4,25.8,24.0,22.7,21.4,20.8.esi-ms:156[m+h]+,[α]
d20
=+10.3
°
(c=1.0in chcl3)。
[0053]
经计算,上述实施例的产率为50.6%,手性薄荷胺的纯度为95%。
[0054]
实施例3
[0055]
本实施例提供一种d-新薄荷胺的制备方法,包括如下步骤:
[0056]
步骤1,3e的合成。称取2.5g(0.016mol)l-型薄荷醇,和12.6g(0.048mol)三苯基膦于100ml反应瓶中,加入10ml四氢呋喃溶解,冰浴搅拌下缓慢滴加8.4ml(0.048mol)偶氮二甲酸二乙酯,反应半小时后加入2.5g(0.024mol)叠氮磷酸二苯酯,并将反应液转移至室温反应8-12h,tlc监测反应完全。反应液经浓缩得粗品,通过硅胶柱层析(pe:ea=40:1,v/v)纯化得到式3e的d-新薄荷叠氮中间体;
[0057]
步骤2、4c的合成。称取1.0g(0.0056mol)d-新薄荷叠氮中间体,和0.15g钯碳加入反应瓶中,加入甲醇溶解,在1-2个大气压的氢气下反应24h,tlc监测反应完全。将反应液用硅藻土过滤,甲醇洗涤3到4次,合并甲醇浓缩后得到无色液体即为式(4c)的d-新薄荷胺。
[0058]
经计算,上述实施例的产率为38.0%,手性薄荷胺的纯度为95%。
[0059]
为了确认合成的手性薄荷胺绝对构型是否正确,我们选择用电子圆二色谱(ecd)法,对比计算和实验中两个对映异构体的ecd图谱的相似程度,来进行构型的确认。由于ecd测试要求手性化合物具有紫外吸收,而最终产物薄荷胺结构中不存在发色团,难以满足这一需求,因此选择肼解前一步的化合物3a以及3a的对映异构体3b(分子结构中存在苯环,见
图1)作为构型分析的对象,其中3b通过其他方式制得,用于辅助判断3a及其后续4a的绝对构型。
[0060]
使用jasco j-810圆二色光谱仪在室温下记录实验ecd光谱,溶剂为色谱级乙醇,浓度控制在0.2mg/ml-0.5mg/ml,以100nm/min的扫描速度记录光谱,响应时间0.5s,累积至少三次扫描,并采用相同溶剂和测试条件对背景进行校正,获得化合物的实验ecd谱。
[0061]
以化合物3a为例,使用discovery studio 4.0分析化合物3a的构象,获得合理的低能构象集,之后使用gaussian 09程序包,通过密度泛函理论(dft)在b3lyp/6-31g(d)水平对这些构象进一步优化,包括几何优化和乙醇的溶剂效应。然后用含时密度泛函理论(tddft)在相同的基组下对优化后的构象进行能量计算。根据计算获得的相关数据,用specdis将其拟合为高斯型曲线,并添加到每个构象的计算ecd光谱中,得到玻尔兹曼加权平均的ecd谱图,最后,通过实验获得的紫外-可见光谱的最大吸收峰对计算ecd谱进行位移值的校正,得到了化合物3a理论的ecd谱。
[0062]
如图1所示,我们用origin 8.5将化合物3a的计算ecd光谱与实验的3a、3b ecd光谱进行比较,3a和3b的实验光谱图呈较为明显的对称形状,可以认为是一对对映异构体,而3a的计算ecd谱与3a的实验光谱在波长和cotton效应方面基本一致,在210nm处存在正性cotton效应,在235nm处存在负性cotton效应,在290nm附近计算ecd光谱存在负性cotton效应,而实验光谱图并没有明显的cotton效应存在,这可能是因为化合物手性碳远离发色团导致的。在肼解反应中一般不会出现手性构型的变化,4a的构型与3a一致,所以可以确认化合物4a的绝对构型为(1r,2r,5r),同理,在同一反应体系下,4c的绝对构型为(1s,2r,5r)。
[0063]
综上所述,本发明以l-薄荷醇为原料,通过一到两次mitsunobu反应合成了两种构型的手性薄荷胺,该合成方法路线新颖,实验操作简单,为新型清凉剂的发现和催化不对称反应提供了合适的工具分子。
[0064]
本领域的技术人员应理解,以上所述仅为本发明的若干个具体实施方式,而不是全部实施例。应当指出,对于本领域的普通技术人员来说,还可以做出许多变形和改进,所有未超出权利要求所述的变形或改进均应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1