一种细菌多重PCR检测引物组合及其应用的制作方法
一种细菌多重pcr检测引物组合及其应用
技术领域
1.本技术涉及微生物检测的技术领域,更具体地说,涉及一种细菌多重pcr检测引物组合及其应用。
背景技术:
2.实验动物的微生物感染不仅对实验动物本身的生长发育、生产及使用产生了重要影响,而且对实验动物从业人员和环境亦造成严重威胁。尤其是实验动物生产机构,无论是从实验动物质量控制还是生物安全的角度,都要投入大量精力对实验动物的微生物进行严格控制。
3.目前对中国实验动物粪便中常见的病原菌,比如金黄色葡萄球菌、绿脓杆菌以及肺炎克雷伯杆菌等进行检测,均依据中国实验动物国家标准gb14922.2-2011中推荐的检测方法进行操作,该方法采用传统的分离培养结合生化鉴定,根据细菌的生物学特性、表型特征进行鉴定。虽然该方法经典、准确,但是存在操作繁琐、试验周期长、效率较低等缺陷,很难做到标准化,与检测试剂、检测程序、硬件条件及检测人员的业务水平和主观判定均有密切联系,易造成各检测批次间检测结果的差异。一些实验动物生产单位拥有众多品系动物,并对生产的实验动物种群进行严格的微生物监控,但是存在着检测周期长、成本高、效率低等问题。
4.pcr技术因具有敏感、快速等优点已被广泛应用于微生物检测。当前国外众多知名的实验动物机构已将分子生物学技术成熟运用于质量监测,近年来中国科研工作者已针对实验动物细菌检测研制开发了不少分子生物学方法,但目前为止中国实验动物微生物检测国标尚未采用该方法。
5.因此,迫切需要开发和建立针对动物微生物感染的快速、可靠的新型监测手段。
技术实现要素:
6.为了对动物的微生物感染情况进行快速、准确、低成本检测,本技术提供一种细菌多重pcr检测引物组合及其应用。本技术提供的引物组合具有良好的特异性与灵敏度,实现了对多样本、多种类细菌的特异、快速、高效检测,并且通过非活体取材,降低了对动物的伤害,具有重要的应用价值。
7.第一方面,本技术提供一种细菌多重pcr检测引物组合,采用如下的技术方案:
8.一种细菌多重pcr检测引物组合,所述细菌多重pcr检测引物组合包括分别对绿脓杆菌、金黄色葡萄球菌、肺炎克雷伯杆菌、奇异变形杆菌、螺旋杆菌和沙门氏菌进行多重pcr检测的引物组合;
9.其中,对所述绿脓杆菌进行多重pcr检测的引物组合包括分别对lasi基因、eta基因和pa基因进行pcr检测的引物中的任意一种或至少两种的组合;
10.对所述lasi基因进行pcr检测的引物包括seq id no.1~2所示的核苷酸序列。
11.优选地,对所述eta基因进行pcr检测的引物包括seq id no.3~4所示的核苷酸序
列。
12.优选地,对所述pa基因进行pcr检测的引物包括seq id no.5~6所示的核苷酸序列。
13.优选地,对所述金黄色葡萄球菌进行多重pcr检测的引物组合包括分别对nuc1基因和/或clfb基因进行pcr检测的引物。
14.优选地,对所述nuc1基因进行pcr检测的引物包括seq id no.7~8所示的核苷酸序列。
15.优选地,对所述clfb基因进行pcr检测的引物包括seq id no.9~10所示的核苷酸序列。
16.优选地,对所述肺炎克雷伯杆菌进行多重pcr检测的引物组合包括分别对kp基因和/或khe基因进行pcr检测的引物。
17.优选地,对所述kp基因进行pcr检测的引物包括seq id no.11~12所示的核苷酸序列。
18.优选地,对所述khe基因进行pcr检测的引物包括seq id no.13~14所示的核苷酸序列。
19.优选地,对所述奇异变形杆菌进行多重pcr检测的引物组合包括分别对urer基因和/或tuf基因进行pcr检测的引物。
20.优选地,对所述urer基因进行pcr检测的引物包括seq id no.15~16所示的核苷酸序列。
21.优选地,对所述tuf基因进行pcr检测的引物包括seq id no.17~18所示的核苷酸序列。
22.优选地,对所述螺旋杆菌进行多重pcr检测的引物组合包括分别对helicobacter和/或bilis进行pcr检测的引物。
23.优选地,对所述helicobacter进行pcr检测的引物包括seq id no.19~20所示的核苷酸序列。
24.优选地,对所述bilis进行pcr检测的引物包括seq id no.21~22所示的核苷酸序列。
25.优选地,对所述沙门氏菌进行多重pcr检测的引物组合包括分别对inva基因和/或ps2基因进行pcr检测的引物。
26.优选地,对所述inva基因进行pcr检测的引物包括seq id no.23~24所示的核苷酸序列。
27.优选地,对所述ps2基因进行pcr检测的引物包括seq id no.25~26所示的核苷酸序列。
28.本技术中,通过对各种病原菌的特异性检测引物进行优化,显著提高了扩增效率,同时简化了细菌鉴定的操作步骤,克服了传统培养法耗时、费力等缺点,可通过一次操作完成多种病原体的同时检测,达到了快速、高效、灵敏、特异的目的。同时也避免了样本数量小、检测范围局限所导致的漏检现象。
29.seq id no.1:atgatcgtacaaattggtcgg(lasi-f)。
30.seq id no.2:gtcatgaaaccgccagtc(lasi-r)。
31.seq id no.3:ttccatggcgagttgct(eta-f)。
32.seq id no.4:cgggcgatctggaaaa(eta-r)。
33.seq id no.5:gcacccgcaacgcatcaa(pa-f)。
34.seq id no.6:cctggaaaggctccgaatagtg(pa-r)。
35.seq id no.7:cacctgaaacaaagcatcctaa(nuc1-f)。
36.seq id no.8:tatacgctaagccacgtccat(nuc1-r)。
37.seq id no.9:acatcagtaatagtagggggcaac(clfb-f)。
38.seq id no.10:ttcgcactgtttgtgtttgcac(clfb-r)。
39.seq id no.11:tggcccgcgccagggttcgaaa(kp-f)。
40.seq id no.12:gatgtcgtcatcgttgatgcccag(kp-r)。
41.seq id no.13:tgattgcattcgccactgg(khe-f)。
42.seq id no.14:ggtcaacccaacgatcctg(khe-r)。
43.seq id no.15:ggtgagatttgtattaatgg(urer-f)。
44.seq id no.16:ataatctggaagatgacgag(urer-r)。
45.seq id no.17:aaattgttgaattagcagaagca(tuf-f)。
46.seq id no.18:gcgattgggtggatcagttc(tuf-r)。
47.seq id no.19:ctatgacgggtatccggc(helicobacter-f)。
48.seq id no.20:ctcacgacacgagctgac(helicobacter-r)。
49.seq id no.21:ctatgacgggtatccggc(bilis-f)。
50.seq id no.22:attccacctacctctccca(bilis-r)。
51.seq id no.23:tttacggtctattttgatttg(inva-f)。
52.seq id no.24:atatgctccacaaggttaatg(inva-r)。
53.seq id no.25:tgtcaccgtggtccagttta(ps2-f)。
54.seq id no.26:cgacaagaccatcaccaatg(ps2-r)。
55.本技术中,上述检测引物组合可以单独使用,对某一种病原菌进行检测与鉴定;多种检测引物组合也可以联合使用,对一种病原菌的多个基因或多种病原菌进行联合检测。
56.第二方面,本技术提供一种细菌多重pcr检测试剂盒,采用如下技术方案:
57.一种细菌多重pcr检测试剂盒,所述细菌多重pcr检测试剂盒包括第一方面所述的细菌多重pcr检测引物组合。
58.优选地,所述细菌多重pcr检测试剂盒还包括扩增试剂、样本处理试剂、dna提取试剂或阳性对照中的任意一种或至少两种的组合。
59.优选地,所述阳性对照包括绿脓杆菌、金黄色葡萄球菌、肺炎克雷伯杆菌、奇异变形杆菌、螺旋杆菌和沙门氏菌的dna。
60.本技术中,通过进一步优化pcr的反应体系,提高了扩增反应的扩增效率,并增加了扩增特异性,进一步保证了检测结果的可信度。
61.本技术中,通过优化单一细菌的多重引物鉴定体系,提高其特异性和稳定性;通过优化多种细菌的引物鉴定体系,提高其特异、稳定及扩增效率,同时减轻扩增过程中不同引物组合之间的竞争及干扰情况。
62.第三方面,本技术提供一种细菌多重pcr检测的方法,采用如下技术方案:
63.一种细菌多重pcr检测的方法,所述细菌多重pcr检测的方法包括使用第一方面所述的细菌多重pcr检测引物组合,或第二方面所述的细菌多重pcr检测试剂盒,对样本进行检测。
64.优选地,所述细菌多重pcr检测的方法包括以下步骤:
65.对样本进行处理;
66.提取处理后的样本的dna;
67.使用第一方面所述的细菌多重pcr检测引物组合,或第二方面所述的细菌多重pcr检测试剂盒,对样本的dna进行检测。
68.本技术中,所述对样本进行处理可以采用以下方式:
69.取新鲜样本,接种到培养基中,35~37℃培养24-36h,备用;
70.取培养上清液150~200μl,10000~12000rpm离心3~5min,弃上清液,加入双蒸水150~200μl,反复吹洗3~5次,10000~12000rpm离心3~5min,清洗2~3次,再加入双蒸水150~200μl。
71.本技术中,可以通过直接加热煮沸的方式,获取处理后的样本的dna。
72.本技术中,用于检测所述螺旋杆菌的样本直接进行加热煮沸处理。
73.其中,所述加热煮沸的温度为98~100℃,所述加热煮沸的时间为8~10min。
74.本技术中,也可以通过蛋白酶消化裂解的方式,提取处理后的样本的dna。
75.其中,所述蛋白酶消化裂解的方式可以采用以下步骤:
76.取490~500μl消化缓冲液与9~10μl蛋白酶k(简称pk,浓度为8~10mg/ml)混合,置于53~58℃的恒温振荡器中孵育过夜;
77.室温下10000~12000rpm离心8~15min,吸取280~300μl上清置于新的离心管中;
78.加入280~300μl异丙醇,来回颠倒离心管使其混合均匀,可见白色絮状dna沉淀;
79.室温下10000~12000rpm离心8~15min,去上清,向沉淀dna中加入100~120μl溶解液溶解,得到样本的dna。
80.优选地,所述方法还包括对阳性对照进行检测的步骤。
81.优选地,所述检测的反应程序包括:预变性;循环延伸;循环外延伸。
82.本技术中,所述预变性的温度为93~98℃,所述预变性的时间为2~5min。
83.本技术中,所述循环扩增可以包括如下程序:
84.变性:93~98℃,15~40s;
85.退火:退火温度(根据使用的引物进行实际选择),15~40s;
86.延伸:70~75℃,延伸时间(按照10~20bp/s的延伸速度,根据使用的引物进行实际确定)。
87.本技术中,所述循环扩增的次数为30~40次。
88.本技术中,所述循环外延伸的温度为70~75℃,所述循环外延伸的时间为5~10min。
89.优选地,所述样本的类型包括粪便、回盲部内容物、肛拭子、病灶组织、病灶组织浓汁和病灶组织分泌物中的任意一种或至少两种的组合。
90.本技术中,可检测样本的类型多,并且避免了活体取样,降低了对动物本身的伤害。对于数量较少的稀有动物品种,这种检测方式更为重要,既保留了群体又对菌群进行了
监测,为动物病原菌的快速检测和流行病学调查提供了一种快速而有效的手段,对于提升实验动物细菌检测能力奠定了基础。
91.第四方面,本技术提供第一方面所述的细菌多重pcr检测引物组合和/或第二方面所述的细菌多重pcr检测试剂盒在制备细菌多重pcr检测产品中的应用。
92.综上所述,本技术具有以下有益效果:
93.(1)本技术所述的检测引物组合具有良好的灵敏度及特异性,可根据具体的检测需求单独或联合使用,灵活性好,容易操作;
94.(2)本技术通过对检测反应的扩增体系及检测程序进行参数优化,进一步提高了扩增效率及扩增的特异性,准确度高,相比于传统的生化鉴定,具有效率高、检测周期短、成本低且易判定的优势;
95.(3)本技术对多种类型的样本均可进行有效检测,采样量大且广,并且可以避免对动物造成伤害,样本的处理方式也较为方便、简单、低成本,为相关产品的推广与使用创造了条件。
附图说明
96.图1为实施例1中样本在nac液体培养基中的培养结果图片。
97.图2为实施例1中样本在nac琼脂平板上的培养结果图片。
98.图3为实施例1中样本培养菌落的革兰氏染色结果图片(放大倍数=1000
×
)。
99.图4为实施例1中使用pcr检测引物的扩增结果图片(图中,泳道m:标准dna分子量marker dl2000;泳道-:阴性对照;泳道1:使用对lasi基因进行pcr检测的引物的扩增结果;泳道2:使用对eta基因进行pcr检测的引物的扩增结果;泳道3:使用对pa基因进行pcr检测的引物的扩增结果;泳道4:联合使用对lasi基因、eta基因和pa基因进行pcr检测的引物的扩增结果)。
100.图5为实施例2中肺炎克雷伯杆菌在dhl琼脂培养基上的培养结果图片。
101.图6为实施例2中样本培养菌落的革兰氏染色结果图片(放大倍数=1000
×
)。
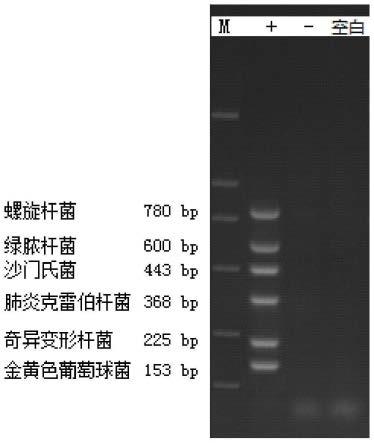
102.图7为实施例2中使用pcr检测引物的扩增结果图片(图中,泳道m:标准dna分子量marker dl2000;泳道+:阳性对照;泳道-:阴性对照;泳道1、2、3、4、5、9、10、11、12、13、14、17、18、19、20、30、32、34、35、39、42和44:编号为1、2、3、4、5、9、10、11、12、13、14、17、18、19、20、30、32、34、35、39、42和44的菌落,联合使用对kp基因和khe基因进行pcr检测的引物的扩增结果)。
103.图8为实施例3中奇异变形杆菌在血琼脂平板上的培养结果图片。
104.图9为实施例3中样本培养菌落的革兰氏染色结果图片(放大倍数=1000
×
)。
105.图10为实施例3中使用pcr检测引物的扩增结果图片(图中,泳道m:标准dna分子量marker dl2000;泳道-:阴性对照;泳道73和78:使用对urer基因进行pcr检测的引物的扩增结果;泳道61和62:使用对tuf基因进行pcr检测的引物的扩增结果)。
106.图11为实施例4中沙门氏菌在dhl琼脂平板上的培养结果图片。
107.图12为实施例4中样本培养菌落的革兰氏染色结果图片(放大倍数=1000
×
)。
108.图13为实施例4中使用pcr检测引物的扩增结果图片(图中,泳道m:标准dna分子量marker dl2000;泳道-:阴性对照;泳道1:使用对inva基因进行pcr检测的引物的扩增结果;
泳道2:使用对ps2基因进行pcr检测的引物的扩增结果)。
109.图14为实施例5中金黄色葡萄球菌在高盐甘露醇琼脂平板上的培养结果图片。
110.图15为实施例5中使用pcr检测引物的扩增结果图片(图中,泳道m:标准dna分子量marker dl2000;泳道+:阳性对照;泳道1:联合使用对nuc1基因和clfb基因进行pcr检测的引物的扩增结果)。
111.图16为实施例6中采用蛋白酶消化裂解获取处理后样本的dna的扩增结果图片(图中,泳道m:标准dna分子量marker dl2000;泳道+:阳性对照;泳道-:阴性对照;泳道1、2、3、9、10、11、12、13、14、17、18、19、20、30、32、34、35、39、42和44:编号为1、2、3、9、10、11、12、13、14、17、18、19、20、30、32、34、35、39、42和44的菌落,联合使用对helicobacter和bilis进行pcr检测的引物的扩增结果)。
112.图17为实施例6中采用直接加热煮沸获取处理后样本的dna的扩增结果图片(图中,泳道m:标准dna分子量marker dl2000;泳道+:阳性对照;泳道44、46、47、48、53和61:编号为44、46、47、48、53和61的菌落,联合使用对helicobacter和bilis进行pcr检测的引物的扩增结果)。
113.图18为实施例7中联合使用6组pcr检测引物的扩增结果图片(图中,泳道m:标准dna分子量marker dl2000;泳道+:阳性样本联合使用6组引物的扩增结果;泳道-:阴性对照;泳道空白:空白对照)。
具体实施方式
114.本技术提供一种细菌多重pcr检测引物组合,所述细菌多重pcr检测引物组合包括分别对绿脓杆菌、金黄色葡萄球菌、肺炎克雷伯杆菌、奇异变形杆菌、螺旋杆菌和沙门氏菌进行多重pcr检测的引物组合;
115.关于对绿脓杆菌的检测:进行多重pcr检测的引物组合包括分别对lasi基因、eta基因和pa基因进行pcr检测的引物。
116.具体地,对所述lasi基因进行pcr检测的引物为seq id no.1~2所示的核苷酸序列。
117.具体地,对所述eta基因进行pcr检测的引物为seq id no.3~4所示的核苷酸序列。
118.具体地,对所述pa基因进行pcr检测的引物为seq id no.5~6所示的核苷酸序列。
119.关于对金黄色葡萄球菌的检测:进行多重pcr检测的引物组合包括分别对nuc1基因和clfb基因进行pcr检测的引物。
120.具体地,对所述nuc1基因进行pcr检测的引物为seq id no.7~8所示的核苷酸序列。
121.具体地,对所述clfb基因进行pcr检测的引物为seq id no.9~10所示的核苷酸序列。
122.关于对肺炎克雷伯杆菌的检测:进行多重pcr检测的引物组合包括分别对kp基因和khe基因进行pcr检测的引物。
123.具体地,对所述kp基因进行pcr检测的引物为seq id no.11~12所示的核苷酸序列。
124.具体地,对所述khe基因进行pcr检测的引物为seq id no.13~14所示的核苷酸序列。
125.关于对奇异变形杆菌的检测:进行多重pcr检测的引物组合包括分别对urer基因和tuf基因进行pcr检测的引物。
126.具体地,对所述urer基因进行pcr检测的引物为seq id no.15~16所示的核苷酸序列。
127.具体地,对所述tuf基因进行pcr检测的引物为seq id no.17~18所示的核苷酸序列。
128.关于对螺旋杆菌的检测:进行多重pcr检测的引物组合包括分别对helicobacter和bilis进行pcr检测的引物。
129.具体地,对所述helicobacter进行pcr检测的引物为seq id no.19~20所示的核苷酸序列。
130.具体地,对所述bilis进行pcr检测的引物为seq id no.21~22所示的核苷酸序列。
131.关于对沙门氏菌的检测:进行多重pcr检测的引物组合包括分别对inva基因和ps2基因进行pcr检测的引物。
132.具体的,对所述inva基因进行pcr检测的引物为seq id no.23~24所示的核苷酸序列。
133.具体的,对所述ps2基因进行pcr检测的引物为seq id no.25~26所示的核苷酸序列。
134.本技术中,上述引物的详细信息如表1所示。
135.表1引物详细信息
136.[0137][0138]
本技术还提供了一种细菌多重pcr检测试剂盒,所述细菌多重pcr检测试剂盒包括上述细菌多重pcr检测引物组合、扩增试剂、样本处理试剂、dna提取试剂和阳性对照。
[0139]
其中,所述扩增试剂包括缓冲液、taq酶、dnips以及mg
2+
等成分。
[0140]
所述样本处理试剂包括细菌培养基和缓冲液。
[0141]
所述dna提取试剂包括消化缓冲液、蛋白酶k和异丙醇等试剂。
[0142]
所述阳性对照为绿脓杆菌、金黄色葡萄球菌、肺炎克雷伯杆菌、奇异变形杆菌、螺旋杆菌和沙门氏菌的dna。
[0143]
本技术还提供了一种细菌多重pcr检测的方法,所述细菌多重pcr检测的方法包括以下步骤:
[0144]
(1)对样本进行处理:
[0145]
取新鲜样本,接种到培养基中,35~37℃培养24~36h备用;
[0146]
取培养上清液150~200μl,10000~12000rpm离心3~5min,弃上清液,加入双蒸水150~200μl,反复吹洗3~5次,10000~12000rpm离心3~5min,清洗2~3次,再加入双蒸水150~200μl。
[0147]
(2)提取处理后的样本的dna:
[0148]
通过直接在98~100℃加热煮沸8~10min,获取处理后的样本的dna,
[0149]
或
[0150]
通过蛋白酶消化裂解的方式,提取处理后的样本的dna,步骤如下:
[0151]
取490~500μl消化缓冲液与9~10μl蛋白酶k(浓度为8~10mg/ml)混合,置于53~58℃下恒温振荡器孵育过夜;
[0152]
室温下10000~12000rpm离心8~15min,吸取280~300μl上清置于新的离心管中;
[0153]
加入280~300μl异丙醇,来回颠倒离心管使其混合均匀,可见白色絮状dna沉淀;
[0154]
室温下10000~12000rpm离心8~15min,去上清,向沉淀dna中加入100~120μl溶
解液溶解,得到样本的dna。
[0155]
(3)使用细菌多重pcr检测引物组合和/或细菌多重pcr检测试剂盒,对样本的dna进行检测:
[0156]
所述检测的反应程序包括:
[0157]
预变性:93~98℃,2~5min;
[0158]
循环延伸:
[0159]
93~98℃,15~40s;
[0160]
退火温度(根据使用的引物进行实际选择),15~40s;
[0161]
70~75℃,延伸时间(按照10~20bp/s的延伸速度,根据使用的引物进行实际确定);
[0162]
循环30~40次;
[0163]
循环外延伸:70~75℃,5~10min。
[0164]
其中,样本的类型包括粪便、回盲部内容物、肛拭子、病灶组织、病灶组织浓汁或病灶组织分泌物中的任意一种或至少两种的组合。
[0165]
在检测的过程中,同步使用阳性对照进行检测,保证检测流程的规范性及结果的准确性。
[0166]
以下结合制备例1-6、实施例1-7以及附图1-18对本技术的技术方案作进一步说明。
[0167]
制备例1-6
[0168]
制备例1
[0169]
本制备例制备一种对绿脓杆菌进行多重pcr检测的引物组合,包括对lasi基因、eta基因和pa基因进行pcr检测的引物组合。
[0170]
对所述lasi基因进行pcr检测的引物包括seq id no.1~2所示的核苷酸序列;
[0171]
对所述eta基因进行pcr检测的引物包括seq id no.3~4所示的核苷酸序列;
[0172]
对所述pa基因进行pcr检测的引物包括seq id no.5~6所示的核苷酸序列。
[0173]
其中,上述引物组合通过如下方式进行设计:
[0174]
选取绿脓杆菌标准菌种,并经生化鉴定和16s引物进行测序鉴定;
[0175]
筛选变异性低的绿脓杆菌的特异性基因,分别设计引物,通过扩增验证特异性,并从中选取扩增效果最优的引物组合,最终选择分别对lasi基因、eta基因和pa基因进行pcr检测的引物作为上述对绿脓杆菌进行多重pcr检测的引物组合。
[0176]
制备例2
[0177]
本制备例制备一种对金黄色葡萄球菌进行多重pcr检测的引物组合,包括分别对nuc1基因和clfb基因进行pcr检测的引物组合。
[0178]
对所述nuc1基因进行pcr检测的引物包括seq id no.7~8所示的核苷酸序列;
[0179]
对所述clfb基因进行pcr检测的引物包括seq id no.9~10所示的核苷酸序列。
[0180]
其中,上述引物组合通过如下方式进行设计:
[0181]
选取金黄色葡萄球菌标准菌种,并经生化鉴定和16s引物进行测序鉴定;
[0182]
筛选变异性低的金黄色葡萄球菌的特异性基因,分别设计引物,通过扩增验证特异性,并从中选取扩增效果最优的引物组合,最终选择分别对nuc1基因和clfb基因进行pcr
检测的引物作为上述对金黄色葡萄球菌进行多重pcr检测的引物组合。
[0183]
制备例3
[0184]
本制备例制备一种对肺炎克雷伯杆菌进行多重pcr检测的引物组合,包括分别对kp基因和khe基因进行pcr检测的引物组合。
[0185]
对所述kp基因进行pcr检测的引物包括seq id no.11~12所示的核苷酸序列;
[0186]
对所述khe基因进行pcr检测的引物包括seq id no.13~14所示的核苷酸序列。
[0187]
其中,上述引物组合通过如下方式进行设计:
[0188]
选取肺炎克雷伯杆菌标准菌种,并经生化鉴定和16s引物进行测序鉴定;
[0189]
筛选变异性低的肺炎克雷伯杆菌的特异性基因,分别设计引物,通过扩增验证特异性,并从中选取扩增效果最优的引物组合,最终选择分别对kp基因和khe基因进行pcr检测的引物作为上述对肺炎克雷伯杆菌进行多重pcr检测的引物组合。
[0190]
制备例4
[0191]
本制备例制备一种对奇异变形杆菌进行多重pcr检测的引物组合,包括分别对urer基因和tuf基因进行pcr检测的引物组合。
[0192]
对所述urer基因进行pcr检测的引物包括seq id no.15~16所示的核苷酸序列;
[0193]
对所述tuf基因进行pcr检测的引物包括seq id no.17~18所示的核苷酸序列。
[0194]
其中,上述引物组合通过如下方式进行设计:
[0195]
选取奇异变形杆菌标准菌种,并经生化鉴定和16s引物进行测序鉴定;
[0196]
筛选变异性低的奇异变形杆菌的特异性基因,分别设计引物,通过扩增验证特异性,并从中选取扩增效果最优的引物组合,最终选择分别对urer基因和tuf基因进行pcr检测的引物作为上述对奇异变形杆菌进行多重pcr检测的引物组合。
[0197]
制备例5
[0198]
本制备例制备一种对螺旋杆菌进行多重pcr检测的引物组合,包括分别对helicobacter和bilis进行pcr检测的引物组合。
[0199]
对所述helicobacter进行pcr检测的引物包括seq id no.19~20所示的核苷酸序列;
[0200]
对所述bilis进行pcr检测的引物包括seq id no.21~22所示的核苷酸序列。
[0201]
其中,上述引物组合通过如下方式进行设计:
[0202]
选取螺旋杆菌helicobacter和bilis标准菌种,并经生化鉴定和16s引物进行测序鉴定;
[0203]
筛选变异性低的特异性基因,分别设计引物,通过扩增验证特异性,并从中选取扩增效果最优的引物组合,最终选择分别对helicobacter和bilis进行pcr检测的引物作为上述对螺旋杆菌进行多重pcr检测的引物组合。
[0204]
制备例6
[0205]
本制备例制备一种对沙门氏菌进行多重pcr检测的引物组合,包括分别对inva基因和ps2基因进行pcr检测的引物组合。
[0206]
对所述inva基因进行pcr检测的引物包括seq id no.23~24所示的核苷酸序列;
[0207]
对所述ps2基因进行pcr检测的引物包括seq id no.25~26所示的核苷酸序列。
[0208]
其中,上述引物组合通过如下方式进行设计:
[0209]
选取沙门氏菌标准菌种,并经生化鉴定和16s引物进行测序鉴定;
[0210]
筛选变异性低的沙门氏菌的特异性基因,分别设计引物,通过扩增验证特异性,并从中选取扩增效果最优的引物组合,最终选择对inva基因和ps2基因进行pcr检测的引物作为上述对沙门氏菌进行多重pcr检测的引物组合。
[0211]
实施例1
[0212]
本实施例分别通过生化鉴定和制备例1制备的对绿脓杆菌进行多重pcr检测的引物组合,对样本中的绿脓杆菌进行鉴定。
[0213]
样本采集:采集回盲部内容物、粪便、病灶组织或病灶组织分泌物。
[0214]
生化鉴定
[0215]
原理:绿脓杆菌是革兰氏阴性杆菌,具有产生绿色色素和氧化酶等生化实验特征,因此,可利用选择性培养基培养、菌体检验和生化试验进行鉴定。鉴定结果如图1-3所示。
[0216]
图1为实施例1中样本在nac液体培养基中的培养结果图片。
[0217]
图2为实施例1中样本在nac琼脂平板上的培养结果图片。
[0218]
图3为实施例1中样本培养菌落的革兰氏染色结果图片(放大倍数=1000
×
)。
[0219]
分离培养:将样本接种至nac液体培养基中,置于37℃下培养24h,观察生长情况,对未产生绿色色素的培养液转种至nac琼脂平板,37℃培养24h,观察并进行革兰氏染色。
[0220]
生长特性及菌落特征:绿脓杆菌在nac液体培养基中均匀混浊生长,有菌膜,大部分菌株可在培养液上半部形成绿色色素(参见图1);在nac琼脂平板上形成扁平、边缘不齐呈锯齿状、直径2~3mm的菌落(参见图2),大部分菌落可产生绿色色素而使培养基呈绿色,部分菌株需延长培养时间方可产生色素。革兰氏染色结果显示为革兰氏阴性菌(参见图3),上述结果证明样本中含有绿脓杆菌。
[0221]
多重pcr检测鉴定
[0222]
分别使用对lasi基因(seq id no.1~2)、eta基因(seq id no.3~4)和pa基因(seq id no.5~6)进行pcr检测的引物,并将上述3组引物联合使用,对样本中的绿脓杆菌进行检测鉴定。
[0223]
所述检测鉴定的方法包括以下步骤:
[0224]
(1)对样本进行处理:
[0225]
将样本接种至nac液体培养基中,37℃培养24h;
[0226]
取培养上清液200μl,12000rpm离心5min,弃上清液,加入双蒸水200μl,反复吹洗5次,12000rpm离心5min,按照上述操作反复清洗2次,再加入200μl双蒸水。
[0227]
(2)提取处理后的样本的dna:
[0228]
取500μl消化缓冲液与7.5μl蛋白酶k(10mg/ml,北京百瑞极生物科技有限公司)混合,置于55℃的恒温振荡器中,消化过夜裂解;其中,消化缓冲液的制备及使用方式如表2所示。
[0229]
表2消化缓冲液的配置及使用方式
[0230]
[0231][0232]
室温下12000rpm离心10min,吸取300μl上清置于新的离心管中;
[0233]
加入300μl异丙醇,来回颠倒离心管使其混合均匀,可见白色絮状dna沉淀;
[0234]
室温下12000rpm离心10min,去上清,向沉淀dna中加入100μl溶解液溶解,得到样本的dna。
[0235]
(3)使用pcr检测引物组合对样本的dna进行检测:
[0236]
在本技术中,pcr扩增反应采用以下反应体系:
[0237]
各组分的加入量为:模板dna 1μl;上游引物0.5μl;下游引物0.5μl;mix 12.5μl;水10.5μl;总体积25μl。
[0238]
上述反应体系中,mix为购自天根生化科技有限公司的商品化产品,为包括缓冲液、taq酶、dnips以及mg
2+
等成分的混合物。
[0239]
在本技术中,pcr扩增反应采用以下反应程序:
[0240]
预变性:95℃,3min;
[0241]
循环延伸:
[0242]
95℃,30s;
[0243]
退火温度(根据使用的引物进行实际选择),30s;
[0244]
72℃,延伸时间(按照10~20bp/s的延伸速度,根据使用的引物进行实际确定);
[0245]
循环35次;
[0246]
循环外延伸:72℃,5min。
[0247]
扩增得到的产物在4℃下保存。
[0248]
(4)对扩增产物进行琼脂糖凝胶电泳:
[0249]
称取0.9g琼脂糖,倒入干净的500ml三角瓶中,加入60ml 1
×
tae缓冲液(取tris 242g、冰醋酸57.1ml和0.5mol/l edta(ph8.0)100ml,混合后用蒸馏水定容至1l,使用时稀释50倍),在微波炉中加热2~3min,使其彻底溶化,溶液透明均一。待琼脂糖凝胶冷却到60℃左右时加入5μlgold view(北京金诺利华科技有限公司),摇匀,徐徐倒入插好样品梳的胶盘中。
[0250]
拔掉样品梳,将凝胶没入盛有1
×
tae缓冲液的电泳槽中。从第二个孔开始,用点胶排枪吸取5μl dna样品至凝胶板上的加样孔中,第一个孔加入同样体积的dna marker。
[0251]
加样完毕后,将电泳槽与电源连接,调整电压为200v,电泳10min。
[0252]
将电泳结束后的凝胶放置于紫外成像仪的样品区,拍摄凝胶图像。
[0253]
扩增产物的电泳结果如图4所示。
[0254]
图4为实施例1中使用pcr检测引物的扩增结果图片(图中,泳道m:标准dna分子量marker dl2000;泳道-:阴性对照;泳道1:使用对lasi基因进行pcr检测的引物的扩增结果;泳道2:使用对eta基因进行pcr检测的引物的扩增结果;泳道3:使用对pa基因进行pcr检测的引物的扩增结果;泳道4:联合使用对lasi基因、eta基因和pa基因进行pcr检测的引物的扩增结果)。
[0255]
由图4可以看出,上述3组引物无论是单独使用或是组合使用,均可以进行有效扩增,条带清晰,各组扩增引物之间无相互干扰,表明样本中确实含有绿脓杆菌。检测结果与生化鉴定的结果一致,表明其具有良好的准确性,且具有耗时短、效率高的优势。
[0256]
实施例2
[0257]
本实施例分别通过生化鉴定和制备例3制备的对肺炎克雷伯杆菌进行多重pcr检测的引物组合,对样本中的肺炎克雷伯杆菌进行鉴定。
[0258]
样本采集:采集回盲部内容物或病灶组织。
[0259]
生化鉴定
[0260]
原理:肺炎克雷伯杆菌在培养基上有特定的生长、形态和生理生化特征。鉴定结果如图5-6所示。
[0261]
图5为实施例2中肺炎克雷伯杆菌在dhl琼脂培养基上的培养结果图片。
[0262]
图6为实施例2中样本培养菌落的革兰氏染色结果图片(放大倍数=1000
×
)。
[0263]
分离培养:将样本接种至dhl琼脂平板,置于37℃下培养24h。
[0264]
菌落特征:肺炎克雷伯杆菌在dhl(胆盐硫乳琼脂培养基)琼脂培养基上形成淡粉色菌落,大而隆起,光滑湿润,呈粘液状,相邻菌落易融合成脓汁样(参见图5),接种针挑取时可拉出较长的丝。革兰氏染色结果显示为革兰氏阴性菌(参见图6),上述结果证明样本中含有肺炎克雷伯杆菌。
[0265]
多重pcr检测鉴定
[0266]
将对kp基因(seq id no.11~12)和khe基因(seq id no.13~14)进行pcr检测的2组引物联合使用,对样本中的肺炎克雷伯杆菌进行检测鉴定。
[0267]
其中,提取处理后的样本的dna采用取培养上清液在100℃加热8min的方式,其余的检测鉴定步骤参见实施例1。
[0268]
扩增产物的电泳结果如图7所示。
[0269]
图7为实施例2中使用pcr检测引物的扩增结果图片(图中,泳道m:标准dna分子量marker dl2000;泳道+:阳性对照;泳道-:阴性对照;泳道1、2、3、4、5、9、10、11、12、13、14、17、18、19、20、30、32、34、35、39、42和44:编号为1、2、3、4、5、9、10、11、12、13、14、17、18、19、20、30、32、34、35、39、42和44的菌落,联合使用对kp基因和khe基因进行pcr检测的引物的扩增结果)。
[0270]
由图7可以看出,上述2组引物联合使用,均可对目标基因进行有效扩增,证明样本中含有肺炎克雷伯杆菌,检测结果与生化鉴定的结果一致。
[0271]
实施例3
[0272]
本实施例分别通过生化鉴定和制备例4制备的对奇异变形杆菌进行多重pcr检测的引物组合,对样本中的奇异变形杆菌进行鉴定。
[0273]
样本采集:采集粪便、回盲部内容物、病灶组织或病灶组织浓汁。
[0274]
生化鉴定
[0275]
原理:奇异变形杆菌在培养基上有特定的生长、形态和生理生化特征。鉴定结果如图8-9所示。
[0276]
图8为实施例3中奇异变形杆菌在血琼脂平板上的培养结果图片。
[0277]
图9为实施例3中样本培养菌落的革兰氏染色结果图片(放大倍数=1000
×
)。
[0278]
分离培养:将样本接种至血平板,置于37℃下培养24h。
[0279]
菌落特征:样本在血琼脂平板上37℃培养24h出现溶血现象,在固体培养基上呈扩散生长,形成迁徒生长现象(参见图8)。革兰氏染色结果显示变形杆菌为革兰氏阴性菌,呈明显的多形性,有球形和丝状形,为周鞭毛菌,运动活泼(参见图9)。上述结果证明样本中含有奇异变形杆菌。
[0280]
多重pcr检测鉴定
[0281]
将对urer基因(seq id no.15~16)和tuf基因(seq id no.17~18)进行pcr检测的2组引物联合使用,对样本中的奇异变形杆菌进行检测鉴定。所述检测鉴定的方法参见实施例1。
[0282]
扩增产物的电泳结果参见图10。
[0283]
图10为实施例3中使用pcr检测引物的扩增结果图片(图中,泳道m:标准dna分子量marker dl2000;泳道-:阴性对照;泳道73和78:使用对urer基因进行pcr检测的引物的扩增结果;泳道61和62:使用对tuf基因进行pcr检测的引物的扩增结果)。
[0284]
由图10可以看出,上述2组引物均可对目标片段进行有效扩增,扩增产物条带清晰,大小也与预期相符,表明样本中含有奇异变形杆菌,检测结果与生化鉴定结果一致。
[0285]
实施例4
[0286]
本实施例分别通过生化鉴定和制备例6制备的对沙门氏菌进行多重pcr检测的引物组合,对样本中的沙门氏菌进行鉴定。
[0287]
样本采集:采集回盲部内容物、粪便或肛拭子。
[0288]
生化鉴定
[0289]
原理:沙门氏菌在培养基上有特定的生长、体态和生理生化特征,菌体抗原与相应的抗体结合,产生凝集反应。鉴定结果如图11-12所示。
[0290]
图11为实施例4中沙门氏菌在dhl琼脂平板上的培养结果图片。
[0291]
图12为实施例4中样本培养菌落的革兰氏染色结果图片(放大倍数=1000
×
)。
[0292]
分离培养:直接分离培养,将样本接种至ss或dhl培养基中,置于37℃下培养24h,再增菌分离培养,将已接种的sf增菌液置于37℃培养过夜,转种ss或dhl培养平板,置于相同温度下培养24h。
[0293]
菌落特征:沙门氏菌在dhl琼脂平板上形成2mm左右、无色半透明、表面光滑湿润的菌落,部分菌落带黑心或全黑(参见图11)。革兰氏染色结果显示为革兰氏阴性菌(参见图12),上述结果证明样本中含有沙门氏菌。
[0294]
多重pcr检测鉴定
[0295]
分别使用对inva基因(seq id no.23~24)和ps2基因(seq id no.25~26)进行pcr检测的引物,对样本中的沙门氏菌进行检测鉴定。所述检测鉴定的方法参见实施例1。
[0296]
扩增产物的电泳结果参见图13。
[0297]
图13为实施例4中使用pcr检测引物的扩增结果图片(图中,泳道m:标准dna分子量marker dl2000;泳道-:阴性对照;泳道1:使用对inva基因进行pcr检测的引物的扩增结果;泳道2:使用对ps2基因进行pcr检测的引物的扩增结果)。
[0298]
由图13可以看出,上述2组引物均可以扩增出目标片段,条带清晰,大小也与预期相符,表明样本中含有沙门氏菌,与生化鉴定的结果相符合。将两种方法进行比较,可以明显看出通过pcr进行检测的方法的操作更加简便,效率更高,成本也更低。
[0299]
实施例5
[0300]
本实施例分别通过生化鉴定和制备例2制备的对金黄色葡萄球菌进行多重pcr检测的引物组合,对样本中的金黄色葡萄球菌进行鉴定。
[0301]
样本采集:采集回盲部内容物、病灶组织或病灶分泌物。
[0302]
生化鉴定
[0303]
原理:金黄色葡萄球菌是革兰氏阳性菌,具有特定的菌落形态、β溶血性和生化反应特征。据此可与其他细菌区别。鉴定结果如图14所示。
[0304]
图14为实施例5中金黄色葡萄球菌在高盐甘露醇琼脂平板上的培养结果图片。
[0305]
分离培养:将样本接种至高盐甘露醇(sp)培养基,置于37℃下培养24h。
[0306]
菌落特征:金黄色葡萄球菌在高盐甘露醇培养基上形成1mm左右、凸起、黄色的菌落,菌落周围的培养基由红变成黄色(参见图14),表明样本中含有金黄色葡萄球菌。
[0307]
多重pcr检测鉴定
[0308]
将对nuc1基因(seq id no.7~8)和clfb基因(seq id no.9~10)进行pcr检测的引物联合使用,对样本中的金黄色葡萄球菌进行检测鉴定。所述检测鉴定的方法参见实施例1。
[0309]
扩增产物的电泳结果参见图15。
[0310]
图15为实施例5中使用pcr检测引物的扩增结果图片(图中,泳道m:标准dna分子量marker dl2000;泳道+:阳性对照;泳道1:联合使用对nuc1基因和clfb基因进行pcr检测的引物的扩增结果)。
[0311]
由图15可以看出,上述2组引物联合使用时未产生干扰现象,二者均可对目标基因进行有效扩增,条带清晰且可区分,扩增产物大小也正确无误,表明样本中含有金黄色葡萄球菌,结果与生化鉴定的结果一致,表明其具有良好的准确性。
[0312]
实施例6
[0313]
本实施例分别采用蛋白酶消化裂解(具体操作参见实施例1)和直接加热煮沸(具体操作参见实施例2)的方式获取处理后样本的dna,并将制备例5制备的对helicobacter(seq id no.19~20)和bilis(seq id no.21~22)进行pcr检测的2组引物联合使用,对样本中的螺旋杆菌进行检测鉴定。所述检测鉴定的方法参见实施例1。
[0314]
采用蛋白酶消化裂解获取处理后样本的dna的扩增结果如图16所示,采用直接加热煮沸获取处理后样本的dna的扩增结果如图17所示。
[0315]
图16为实施例6中采用蛋白酶消化裂解获取处理后样本的dna的扩增结果图片(图
中,泳道m:标准dna分子量marker dl2000;泳道+:阳性对照;泳道-:阴性对照;泳道1、2、3、9、10、11、12、13、14、17、18、19、20、30、32、34、35、39、42和44:编号为1、2、3、9、10、11、12、13、14、17、18、19、20、30、32、34、35、39、42和44的菌落,联合使用对helicobacter和bilis进行pcr检测的引物的扩增结果)。
[0316]
图17为实施例6中采用直接加热煮沸获取处理后样本的dna的扩增结果图片(图中,泳道m:标准dna分子量marker dl2000;泳道+:阳性对照;泳道44、46、47、48、53和61:编号为44、46、47、48、53和61的菌落,联合使用对helicobacter和bilis进行pcr检测的引物的扩增结果)。
[0317]
由图16和17可以看出,2组引物联合使用时也均具有良好的扩增效率。阴形对照无扩增条带,阳性对照可以扩增出预期大小的扩增产物,表明样本中含有螺旋杆菌,且检测结果具有良好的特异性,结果准确,特异性好。
[0318]
实施例7
[0319]
本实施例使用制备例1-6分别制备的细菌多重pcr检测引物组合的混合物,以混有绿脓杆菌、金黄色葡萄球菌、肺炎克雷伯杆菌、奇异变形杆菌、螺旋杆菌和沙门氏菌6种细菌的阳性样本作为待测对象,对引物的扩增性能进行验证。
[0320]
所述验证的方法包括以下步骤:
[0321]
(1)对样本进行处理:
[0322]
将样本接种至营养肉汤培养基中,37℃培养24h;
[0323]
取培养上清液200μl,12000rpm离心5min,弃上清液,加入双蒸水200μl,反复吹洗5次,12000rpm离心5min,按照上述操作反复清洗2次,再加入200μl双蒸水。
[0324]
(2)提取处理后的样本的dna:
[0325]
参见实施例1。
[0326]
(3)使用pcr检测引物组合对样本的dna进行检测:
[0327]
参见实施例1。
[0328]
(4)对扩增产物进行琼脂糖凝胶电泳:
[0329]
参见实施例1。
[0330]
扩增产物的电泳结果如图18所示。
[0331]
图18为实施例7中联合使用6组pcr检测引物的扩增结果图片(图中,泳道m:标准dna分子量marker dl2000;泳道+:阳性样本联合使用6组引物的扩增结果;泳道-:阴性对照;泳道空白:空白对照)。
[0332]
由图18可以看出,上述6组引物联合使用时无相互干扰,每一组引物均可进行有效扩增,条带清晰,且条带大小与预期相符。表明上述引物组合具有良好的特异性,可以同时对样本进行检测,大大节省了检测的时间和成本,具有广阔的应用前景。
[0333]
本具体实施例仅仅是对本技术的解释,其并不是对本技术的限制,本领域技术人员在阅读完本说明书后可以根据需要对本实施例做出没有创造性贡献的修改,但只要在本技术的权利要求范围内都受到专利法的保护。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1