一种头孢泊肟酯杂质I的制备方法与流程
一种头孢泊肟酯杂质i的制备方法
技术领域
1.本发明属于有机合成领域,具体涉及一种头孢泊肟酯杂质i的制备方法。
背景技术:
2.头孢泊肟酯(cefpodoxime proxetil,cpdx-pr)系第3代口服头孢菌素,1990年在日本首次上市,其结构特征是头孢骨架7位连有甲氧亚氨噻唑基,3位连有甲氧甲基,4位羧酸上有proxetil基。cpdx-pr为头孢泊肟(cefpodoxime,cpdx)的前体药物,本身无抗菌活性,口服后经肠道吸收,在肠壁被非特异性的酯酶水解成cpdx而发挥抗菌活性。cpdx通过与特异性青霉素结合蛋白1a,1b,2和3的作用,引起异常的细菌细胞壁合成和细胞溶解。cpdx有广谱而强大的抗菌作用,对多数革兰阳性菌、阴性菌均有抑制及杀死作用,并对多种β内酰胺酶具有高度稳定性。广泛应用于呼吸道、泌尿道、妇产科感染性疾病和化脓性中耳炎等的治疗。
3.目前,尚无有关制备头孢泊肟酯杂质i的文献报道。中国药典(2020版)中头孢泊肟酯的质量标准尤其是有关物质项中有了更加严格的规定,其中规定:单个未知杂质峰限度均不得大于0.10%;头孢泊肟酯杂质i单个主峰面积限度不得大于对照溶液两主峰面积之和的0.1 倍 (0.1%)。近年来,随着仿制药一致性评价工作的推进,药物的质量研究工作尤其是杂质的研究变得更加重要。
技术实现要素:
4.本发明的目的是提供一种合成头孢泊肟酯杂质i的方法。该方法操作简单,反应周期短,可为头孢泊肟酯原料药质量控制提供合格的对照品。
5.为了实现上述目的,本发明通过如下技术方案实现:一种头孢泊肟酯杂质i的制备方法,具体合成路线如下:。
6.头孢泊肟酯杂质i的制备方法,包括以下步骤:(1)中间体1的制备氮气保护下,以有机溶媒作为反应溶剂,加入化合物a,然后加入n,n-二异丙基乙
胺,保持温度在-5℃~15℃,滴加氯甲酸异丙酯,反应4~8h;反应完成后,加入水,25℃~30℃搅拌15min,分液,取上层水相,滴加85%甲酸溶液调节ph至2.2~2.5,养晶30min后抽滤,干燥得到中间体1;(2)中间体2的制备取步骤(1)制备的中间体1,加入到四氢呋喃和水的混合物中,保持反应温度在5℃~35℃,加入化合物b,滴加三乙胺,反应3~6h;反应完成后,加入乙酸乙酯,25℃~30℃搅拌15min,分液,取下层水相,滴加0.2mol/l 盐酸溶液调节ph至2.8~3.2,养晶60min后抽滤,干燥得到中间体2;(3)杂质i的制备以极性溶媒作为反应溶剂,氮气保护下,加入中间体2和头孢泊肟酯侧链,保持反应温度为-30℃~10℃,滴加1,8-二氮杂双环[5.4.0]十一碳-7-烯 (dbu),搅拌反应2小时;加入纯化水和乙酸乙酯,搅拌0.5小时,静置,分液,浓缩有机相至基本无液滴出现,加入甲醇和1mol/l 盐酸溶液,将其滴加至水中,滴加结束后,搅拌养晶30min后抽滤,干燥得到头孢泊肟酯杂质i。
[0007]
步骤(1)中,所述的化合物a:有机溶媒:n,n-二异丙基乙胺:氯甲酸异丙酯的用量质量比为1:10~15: 0.5~0.8:0.35~0.55。
[0008]
步骤(1)中,所述的有机溶媒选自二氯甲烷、乙酸乙酯或者甲苯。
[0009]
步骤(2)中,所述的中间体1:中间体b:四氢呋喃:水:三乙胺的质量比为1:0.6~1.0:6~10:5~8:0.35~0.55。
[0010]
步骤(3)中,所述的中间体2:n,n-二甲基甲酰胺:dbu:头孢泊肟酯侧链的质量比为:1:5~15:0.25~0.45:0.45~0.65。
[0011]
步骤(3)中,所述的中间体2:水:乙酸乙酯:甲醇:1mol/l 盐酸溶液的质量比为1:8~15:10 ~20:3~5:0.5~1.5。
[0012]
步骤(3)中,所述的极性溶媒包括但不限于n,n-二甲基甲酰胺、二甲基亚砜或者环丁砜。
[0013]
步骤(3)中,所述的头孢泊肟酯侧链选自1-氯乙基异丙基碳酸酯、1-碘乙基异丙基碳酸酯或两者的混合。
[0014]
本发明的有益效果:是提供一种合成头孢泊肟酯杂质i的方法。该方法操作简单,反应周期短,为头孢泊肟酯原料药质量控制提供合格的对照品。该方法无需过柱纯化,有效缩短了制备周期,提高了工作效率,杂质i纯度在94.0%以上。
附图说明
[0015]
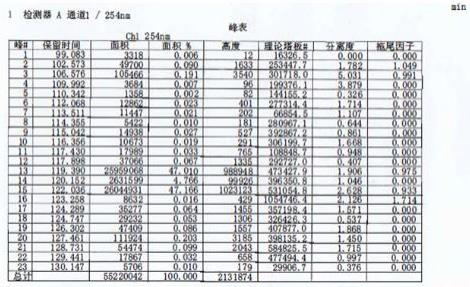
图1为高效液相(hplc)检测的杂质i纯度数据图2为杂质i的核磁共振氢谱图谱图3为杂质i的核磁共振碳谱图谱从图1中可以看出保留时间119.390min和122.036min分别是杂质i
‑ⅰ
和杂质i
‑ⅱ
的峰,两者之和为杂质i的纯度;
具体实施方式
[0016]
下述实例仅用于说明本发明,不用于限制本发明的范围实施例1在氮气保护下,将35.0g ae活性酯(化合物a,100mmol)悬浮于300ml二氯甲烷,加入n,n-二异丙基乙胺20.68g(160 mmol),降温至0℃,向上述母液中滴加氯甲酸异丙酯18.3g(150 mmol);反应约5h,tlc检测,反应完全。加入315ml水,升温至25℃~30℃搅拌约15min,分液,取上层水相,滴加约14.0g 85%甲酸溶液调节ph至2.2~2.5,养晶约30min后抽滤,干燥得到中间体1。
[0017]
取21.0g(50mmol)中间体1,加入175ml四氢呋喃和135ml水,保持反应温度为20℃,加入14.65g(60mmol)化合物b(7-氨基-3-甲氧甲基-3-头孢烯-4-羧酸,7-amca),滴加三乙胺10.12g(100mmol),反应6h;反应完成后,加入140ml乙酸乙酯, 30℃搅拌约15min,分液,取下层水相,滴加约125g 0.2mol/l 盐酸溶液调节ph至2.8~3.2,养晶60min后抽滤,干燥得到中间体222.44g。
[0018]
以130mln,n-二甲基甲酰胺作为反应溶剂,氮气保护下,加入中间体2 15.33g(30 mmol)和1-氯乙基异丙基碳酸酯7.50g(45 mmol),反应温度为0℃,滴加1,8-二氮杂双环[5.4.0]十一碳-7-烯5.48g (dbu,36 mmol),搅拌反应约2小时。反应完成后,加入150ml纯化水和250ml乙酸乙酯,搅拌约0.5小时,静置,分液,浓缩有机相至基本无液滴出现,加入60ml甲醇和15.3g 1mol/l 盐酸溶液,将其滴加至300ml水中,滴加结束后,搅拌养晶约30min后抽滤,干燥得到头孢泊肟酯杂质i6.84g,纯度94.1%。
[0019]
实施例2在氮气保护下,将70.0g ae活性酯(化合物a,200mmol)悬浮于650ml二氯甲烷,加入n,n-二异丙基乙胺43.95g(170 mmol),降温至0℃,向上述母液中滴加氯甲酸异丙酯48.8g(400 mmol);反应5h,tlc检测,反应完全。加入650ml水,升温至25℃~30℃搅拌约15min,分液,取上层水相,滴加约30.0g 85%甲酸溶液调节ph至2.2~2.5,养晶约30min后抽滤,干燥得到中间体145.75g。
[0020]
取42.0g(100mmol)中间体1,加入175ml四氢呋喃和135ml水,保持反应温度为20℃,加入29.30g(120mmol)化合物b(7-氨基-3-甲氧甲基-3-头孢烯-4-羧酸,7-amca),滴加三乙胺20.24g(200mmol),反应约6h;反应完成后,加入300ml乙酸乙酯, 30℃搅拌约15min,分液,取下层水相,滴加约250g 0.2mol/l 盐酸溶液调节ph至2.8~3.2,养晶约60min后抽滤,干燥得到中间体243.21g。
[0021]
以250mln,n-二甲基甲酰胺作为反应溶剂,氮气保护下,加入中间体2 30.66g(60 mmol)和1-碘乙基异丙基碳酸酯23.22g(90 mmol),反应温度为0℃,滴加1,8-二氮杂双环[5.4.0]十一碳-7-烯10.96g (dbu,72 mmol),搅拌反应约2小时。反应完成后,加入300ml纯化水和500ml乙酸乙酯,搅拌约0.5小时,静置,分液,浓缩有机相至基本无液滴出现,加入130ml甲醇和30.60g 1mol/l 盐酸溶液,将其滴加至650ml水中,滴加结束后,搅拌养晶约30min后抽滤,干燥得到头孢泊肟酯杂质i12.55g,纯度94.3%。
[0022]
实施例3在氮气保护下,将52.5g ae活性酯(化合物a,150mmol)悬浮于700ml乙酸乙酯,加入n,n-二异丙基乙胺31.5g(122 mmol),降温至0℃,向上述母液中滴加氯甲酸异丙酯21.0g
(172 mmol);反应5h,tlc检测,反应完全。加入450ml水,升温至25℃~30℃搅拌约15min,分液,取上层水相,滴加约22.0g 85%甲酸溶液调节ph至2.2~2.5,养晶约30min后抽滤,干燥得到中间体134.33g。
[0023]
取30.0g(71.4mmol)中间体1,加入250ml四氢呋喃和193ml水,保持反应温度为25℃,加入20.95g(85.8mmol)化合物b(7-氨基-3-甲氧甲基-3-头孢烯-4-羧酸,7-amca),滴加三乙胺14.47g(143mmol),反应约6h;反应完成后,加入200ml乙酸乙酯, 30℃搅拌约15min,分液,取下层水相,滴加约180g 0.2mol/l 盐酸溶液调节ph至2.8~3.2,养晶约60min后抽滤,干燥得到中间体231.55g。
[0024]
以170mln,n-二甲基甲酰胺作为反应溶剂,氮气保护下,加入中间体20.0g(39mmol)和1-碘乙基异丙基碳酸酯15.18g(59mmol),反应温度为0℃,滴加1,8-二氮杂双环[5.4.0]十一碳-7-烯7.16g (dbu,47 mmol),搅拌反应约2小时。反应完成后,加入200ml纯化水和350ml乙酸乙酯,搅拌约0.5小时,静置,分液,浓缩有机相至基本无液滴出现,加入100ml甲醇和20.0g 1mol/l 盐酸溶液,将其滴加至450ml水中,滴加结束后,搅拌养晶约30min后抽滤,干燥得到头孢泊肟酯杂质i8.24g,纯度94.8%。
[0025]
实施例4在氮气保护下,将105g ae活性酯(化合物a,300mmol)悬浮于1400ml乙酸乙酯,加入n,n-二异丙基乙胺63.0g(244 mmol),降温至-5℃,向上述母液中滴加氯甲酸异丙酯42.0g(344 mmol);反应6h,tlc检测,反应完全。加入900ml水,升温至25℃~30℃搅拌约15min,分液,取上层水相,滴加约44.0g 85%甲酸溶液调节ph至2.2~2.5,养晶约30min后抽滤,干燥得到中间体1 68.50g。
[0026]
取60.0g(142.8mmol)中间体1,加入500ml四氢呋喃和386ml水,保持反应温度为30℃,加入41.9g(171.6mmol)化合物b(7-氨基-3-甲氧甲基-3-头孢烯-4-羧酸,7-amca),滴加三乙胺28.95g(286mmol),反应约6h;反应完成后,加入400ml乙酸乙酯, 30℃搅拌约15min,分液,取下层水相,滴加约360g 0.2mol/l 盐酸溶液调节ph至2.8~3.2,养晶约60min后抽滤,干燥得到中间体263.0g。
[0027]
以425mln,n-二甲基甲酰胺作为反应溶剂,氮气保护下,加入中间体50.0g(97.5mmol)和1-碘乙基异丙基碳酸酯37.95g(147.5mmol),反应温度为0℃,滴加1,8-二氮杂双环[5.4.0]十一碳-7-烯17.9g (dbu,117.5 mmol),搅拌反应约2小时。反应完成后,加入500ml纯化水和850ml乙酸乙酯,搅拌约0.5小时,静置,分液,浓缩有机相至基本无液滴出现,加入250ml甲醇和50g 1mol/l 盐酸溶液,将其滴加至1100ml水中,滴加结束后,搅拌养晶约30min后抽滤,干燥得到头孢泊肟酯杂质i20.5g,纯度94.3%。
[0028]
实施例5在氮气保护下,将59.50g ae活性酯(化合物a,170mmol)悬浮于500ml二氯甲烷,加入n,n-二异丙基乙胺31.0g(240 mmol),降温至0℃,向上述母液中滴加氯甲酸异丙酯29.8g(244 mmol);反应6h,tlc检测,反应完全。加入500ml水,升温至25℃~30℃搅拌约15min,分液,取上层水相,滴加约24.0g 85%甲酸溶液调节ph至2.2~2.5,养晶约30min后抽滤,干燥得到中间体1。
[0029]
取35.7g(85mmol)中间体1,加入300ml四氢呋喃和220ml水,保持反应温度为20℃,加入24.9g(102mmol)化合物b(7-氨基-3-甲氧甲基-3-头孢烯-4-羧酸,7-amca),滴加三乙
胺16.8g(166mmol),反应6h;反应完成后,加入250ml乙酸乙酯, 30℃搅拌约15min,分液,取下层水相,滴加约212g 0.2mol/l 盐酸溶液调节ph至2.8~3.2,养晶60min后抽滤,干燥得到中间体238.15g。
[0030]
以250mln,n-二甲基甲酰胺作为反应溶剂,氮气保护下,加入中间体2 26.0g(51 mmol)和1-氯乙基异丙基碳酸酯12.67g(45 mmol),反应温度为0℃,滴加1,8-二氮杂双环[5.4.0]十一碳-7-烯9.89g (dbu,65mmol),搅拌反应约2小时。反应完成后,加入270ml纯化水和400ml乙酸乙酯,搅拌约0.5小时,静置,分液,浓缩有机相至基本无液滴出现,加入120ml甲醇和26.0g 1mol/l 盐酸溶液,将其滴加至500ml水中,滴加结束后,搅拌养晶约30min后抽滤,干燥得到头孢泊肟酯杂质i 11.63g,纯度94.5%。
[0031]
实施例6在氮气保护下,将87.5g ae活性酯(化合物a,250mmol)悬浮于1200ml乙酸乙酯,加入n,n-二异丙基乙胺61.0g(472 mmol),降温至0℃,向上述母液中滴加氯甲酸异丙酯41.8g(342 mmol);反应约5h,tlc检测,反应完全。加入750ml水,升温至25℃~30℃搅拌约15min,分液,取上层水相,滴加约47.8g 85%甲酸溶液调节ph至2.2~2.5,养晶约30min后抽滤,干燥得到中间体1。
[0032]
取52.5g(125mmol)中间体1,加入500ml四氢呋喃和370ml水,保持反应温度为25℃,加入48.9g(200mmol)化合物b(7-氨基-3-甲氧甲基-3-头孢烯-4-羧酸,7-amca),滴加三乙胺24.7g(244mmol),反应6h;反应完成后,加入370ml乙酸乙酯, 30℃搅拌约15min,分液,取下层水相,滴加约378g 0.2mol/l 盐酸溶液调节ph至2.8~3.2,养晶60min后抽滤,干燥得到中间体256.1g。
[0033]
以400mln,n-二甲基甲酰胺作为反应溶剂,氮气保护下,加入中间体2 38.3g(51 mmol)和1-氯乙基异丙基碳酸酯22.7g(136 mmol),反应温度为0℃,滴加1,8-二氮杂双环[5.4.0]十一碳-7-烯15.6g (dbu,102mmol),搅拌反应约2小时。反应完成后,加入400ml纯化水和650ml乙酸乙酯,搅拌约0.5小时,静置,分液,浓缩有机相至基本无液滴出现,加入220ml甲醇和46.0g 1mol/l 盐酸溶液,将其滴加至700ml水中,滴加结束后,搅拌养晶约30min后抽滤,干燥得到头孢泊肟酯杂质i 17.2g,纯度94.2%。
[0034]
实施例7在氮气保护下,将94.5g ae活性酯(化合物a,270mmol)悬浮于1000ml二氯甲烷,加入n,n-二异丙基乙胺69.0g(534 mmol),降温至10℃,向上述母液中滴加氯甲酸异丙酯43.0g(352mmol);反应约6h,tlc检测,反应完全。加入800ml水,升温至25℃~30℃搅拌约15min,分液,取上层水相,滴加约49.8g 85%甲酸溶液调节ph至2.2~2.5,养晶约30min后抽滤,干燥得到中间体1。
[0035]
取56.7g(135mmol)中间体1,加入500ml四氢呋喃和370ml水,保持反应温度为25℃,加入50.9g(208mmol)化合物b(7-氨基-3-甲氧甲基-3-头孢烯-4-羧酸,7-amca),滴加三乙胺26.7g(264mmol),反应6h;反应完成后,加入400ml乙酸乙酯, 30℃搅拌约15min,分液,取下层水相,滴加约400g 0.2mol/l 盐酸溶液调节ph至2.8~3.2,养晶60min后抽滤,干燥得到中间体2 60.3g。
[0036]
以400mln,n-二甲基甲酰胺作为反应溶剂,氮气保护下,加入中间体2 41.0g(80 mmol)和1-氯乙基异丙基碳酸酯24.7g(148 mmol),反应温度为0℃,滴加1,8-二氮杂双环
[5.4.0]十一碳-7-烯16.6g (dbu,109mmol),搅拌反应约2小时。反应完成后,加入450ml纯化水和700ml乙酸乙酯,搅拌约0.5小时,静置,分液,浓缩有机相至基本无液滴出现,加入220ml甲醇和54.0g 1mol/l 盐酸溶液,将其滴加至750ml水中,滴加结束后,搅拌养晶约30min后抽滤,干燥得到头孢泊肟酯杂质i 18.45g,纯度94.5%。
[0037]
根据本发明所公开的内容,本领域技术人员可最大限度地应用本发明。因此,上述优选具体实施方式仅为举例说明,而非以任何方式限制本发明的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1