一种生产2的制作方法
一种生产2
′‑
脱氧胞苷的重组微生物及方法
技术领域
1.本发明属于生物技术领域,具体涉及一种生产2
′‑
脱氧胞苷的重组微生物及方法。
背景技术:
[0002]2′‑
脱氧胞苷是一种以胞嘧啶为碱基的嘧啶2
′‑
脱氧核糖核苷,由胞嘧啶和2
′‑
脱氧核糖组成,分子式为c9h
15
n3o4,分子量为227.22,类白色粉末,易溶于水。2
′‑
脱氧胞苷是一种重要的医药中间体,主要用于化学合成各种药物,如地西他滨(dacogen
tm
,5-氮杂-2
′‑
脱氧胞苷),用于治疗骨髓增生异常综合征(某些血细胞功能失调的一类疾病)和急性髓系白血病等。此外,2
′‑
脱氧胞苷还被用来合成脱氧核苷酸的单体,以及制备寡聚脱氧核苷酸,广泛应用于核酸与基因工程中,如dntp和各类引物等。
[0003]
目前2
′‑
脱氧胞苷主要是化学合成为主:以α-d-2-脱氧核糖和尿嘧啶为起始原料,经过甲苷化、酯化、氯代、亲核取代、高效氨化及水解脱保护等6个步骤,经一次重结晶得到目标物2
′‑
脱氧胞苷。但是化学法生产2
′‑
脱氧胞苷相对繁琐、成本高,且产物得率较低。与化学生产工艺相比,利用微生物生产2
′‑
脱氧胞苷的成本效益较好,近年来逐渐成为热点。例如,lee等人通过化学诱变构建基因工程产氨棒杆菌进行2
′‑
脱氧胞苷的生产(lee yb,baek h,kim sk,hyun hh.et al.deoxycytidine production by metabolically engineered corynebacterium ammoniagenes.j microbiol 49:53
–
57(2011))。此外,kim等人通过敲除大肠杆菌bl21(de3)基因组的deoa、udp、deod、dcd、cdd、coda等一系列基因和过表达基因t4噬菌体来源的nrdbca、yfbr及pyrg等基因进行2
′‑
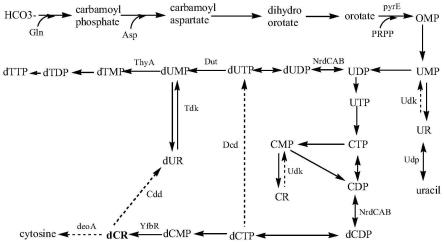
脱氧胞苷的生产(kim,js.,koo,bs.,hyun,hh.et al.deoxycytidine production by a metabolically engineered escherichia coli strain.microb cell fact 14,98(2015))。
[0004]
然而,采用上述文献报道的重组微生物菌株发酵法生产2
′‑
脱氧胞苷产量提高有限,且使用的质粒是中高拷贝,随着发酵时间延长存在质粒不稳定容易丢失的缺陷,此外还存在菌种放大的困难。因此,亟需寻找一种提高微生物发酵生产2
′‑
脱氧胞苷的产量,且易于操作,可应用工业化生产的方法。
技术实现要素:
[0005]
针对以上问题,本发明提供了一种生产2
′‑
脱氧胞苷的重组微生物及方法。本发明意外发现过表达非特异性的5
′‑
脱氧核苷酸酶yfdr(5
′‑
deoxynucleotidase)比文献报道的特异性的dcmp磷酸水解酶yfbr(dcmp phosphohydrolase)更利于提高2
′‑
脱氧胞苷的产量,而且二者联用进行共表达提高2
′‑
脱氧胞苷产量的效果更优。该重组微生物可用于2
′‑
脱氧胞苷的工业化生产,且易于操作,从而达到绿色环保低成本生产2
′‑
脱氧胞苷的目的。
[0006]
术语:
[0007]
1、2
′‑
脱氧胞苷:如无特殊说明,本发明中的英文简称“dcr”指2
′‑
脱氧胞苷,其分子式为c9h
15
n3o4,分子量为227.22,结构式如下式(1)所示。
[0008][0009]
2、重组微生物:如无特殊说明,本发明中的中文名称“重组微生物”指经过人为处理后的微生物;所述的人为处理包括但不限于:基因敲除、基因抑制、基因沉默、基因插入、基因突变、外源表达或过表达基因等本领域所有可以实现基因操作的技术手段。
[0010]
3、宿主菌:如无特殊说明,本发明中的中文名称“宿主菌”指待进行人为处理的微生物。
[0011]
4、种子液:如无特殊说明,本发明中的中文名称“种子液”指经微生物经试管斜面活化、扁平或摇瓶培养或种子罐逐级扩大培养后,获得的含有一定微生物数量,活力、质量较优的微生物培养液。
[0012]
5、高产:如无特殊说明,本发明中的“高产”指2
′‑
脱氧胞苷产量高于亲本微生物。
[0013]
6、低产:如无特殊说明,本发明中的“低产”指2
′‑
脱氧胞苷产量低于亲本微生物。
[0014]
7、外源表达或过表达:在相对于本发明使用时应被广义地认为包括在相同条件下与亲本微生物的一种或多种蛋白质(包括编码一种或多种蛋白质的一种或多种核酸)表达水平相比所述蛋白质表达(包括核酸表达)的任何增加。不应被认为意指所述蛋白质(或核酸)以任何具体水平表达。
[0015]
8、亲本微生物:是本发明的微生物所来源于的微生物。本发明的微生物可以通过任何方法例如人工或天然选择、突变或基因重组而产生。亲本微生物可以是天然存在的微生物(即野生型微生物)或先前曾被修饰过,但是不生产或不过度生产2
′‑
脱氧胞苷的微生物。
[0016]
9、载体:应被广义地认为包括适合用作媒介将遗传物质转移到细胞中的任何核酸(包括dna和rna),包括质粒、病毒(包括噬菌体)、粘粒和人工染色体。载体可以包括一种或多种调节元件、复制起点、多克隆位点和/或可选择标记物。
[0017]
10、关于蛋白和基因名称的约定:本发明中涉及的英文蛋白名称均为首字母大写正体;本发明中涉及的英文基因名称均为首字母小写正体。
[0018]
为了实现上述发明目的,本发明的技术方案如下:
[0019]
一方面,本发明提供了5
′‑
脱氧核苷酸酶和/或dcmp磷酸水解酶或其编码基因在生产2
′‑
脱氧胞苷中的应用。
[0020]
具体地,所述的5
′‑
脱氧核苷酸酶为yfdr,所述的dcmp磷酸水解酶为yfbr。
[0021]
另一方面,本发明提供了一种重组微生物,所述的重组微生物包括以下特征:5
′‑
脱氧核苷酸酶和/或dcmp磷酸水解酶的表达量或活性提高,和/或,5
′‑
脱氧核苷酸酶和/或dcmp磷酸水解酶的编码基因增加、拷贝数或活力提高;所述的重组微生物用于生产2
′‑
脱氧胞苷。
[0022]
具体地,所述的5
′‑
脱氧核苷酸酶为yfdr,所述的dcmp磷酸水解酶为yfbr。
[0023]
具体地,所述的重组微生物还包括以下特征:核糖核苷二磷酸还原酶(ribonucleoside-diphosphate reductase)及硫氧还蛋白的含量或活性提高,和/或,核糖核苷二磷酸还原酶及硫氧还蛋白的编码基因增加、拷贝数或活力提高。
[0024]
进一步具体地,所述的核糖核苷二磷酸还原酶为t4噬菌体来源的核糖二磷酸还原酶nrdb、nrda,所述的硫氧还蛋白为nrdc。
[0025]
具体地,所述的微生物包括但不限于大肠杆菌(escherichia coli)、芽孢杆菌(bacillus)、谷氨酸棒杆菌(corynebacterium glutamicum)、沙门氏菌(salmonella)和酵母菌(yeast)。
[0026]
又一方面,本发明提供了上述重组微生物在生产2
′‑
脱氧胞苷中的应用。
[0027]
又一方面,本发明提供了一种生产2
′‑
脱氧胞苷的方法,所述的方法包括利用上述重组微生物发酵生产2
′‑
脱氧胞苷或利用上述重组微生物全细胞产物催化底物生产2
′‑
脱氧胞苷。
[0028]
具体地,所述的全细胞产物包括但不限于:培养液、细胞裂解液、细胞裂解液的上清部分、细胞裂解液的沉淀部分等。需要说明的是,在未来的技术发展中对于全细胞产物的操作方法及其具体产物类型的新增技术由于未脱离本发明的发明点,也应当在本发明的上述要求保护的范围之内。
[0029]
具体地,所述的方法包括:
[0030]
将上述重组微生物活化后制备种子液;
[0031]
采用摇瓶发酵:按照1-5%接种量接种到发酵培养基中,37℃摇床250rpm培养3-5h,添加终浓度为1mm的iptg,调节摇床温度至34℃,发酵周期为18-22h。
[0032]
进一步具体地,所述的接种量为1-2%。
[0033]
进一步具体地,所述的发酵培养基成分为:每升培养基中葡萄糖20-40g,5n-5倍的盐溶液180-220ml,tm2溶液0.5-1.5ml,柠檬酸铁8-12mg,七水硫酸镁240-250mg,氯化钙105-115mg,硫胺素0.5-1.5μg,以无菌去离子水定容至1l;其中5n-5倍的盐溶液为十二水合磷酸氢二钠75.6g,磷酸二氢钾15g,氯化钠2.5g,氯化铵25g,以无菌去离子水定容至1l;tm3溶液为四水氯化锌2.0g,六水氯化钙2.0g,两水钼酸钠2.0g,五水硫酸铜1.9g,硼酸0.5g,盐酸100ml,去离子水定容至1l。
[0034]
进一步具体地,所述的发酵周期为20h。
[0035]
进一步具体地,所述的发酵培养基成分为:每升培养基中葡萄糖30g,5n-5倍的盐溶液200ml,tm2溶液1ml,柠檬酸铁10mg,七水硫酸镁246mg,氯化钙111mg,硫胺素1μg,以无菌去离子水定容至1l。
[0036]
与现有技术相比,本发明的积极和有益效果在于:
[0037]
(1)本发明意外发现,实际生产过程中,过表达非特异性的5
′‑
脱氧核苷酸酶yfdr(5
′‑
deoxynucleotidase)比文献报道的特异性的dcmp磷酸水解酶yfbr(dcmp phosphohydrolase)更利于提高2
′‑
脱氧胞苷的产量,克服了现有技术的技术偏见,且yfdr与yfbr二者联用进行共表达提高2
′‑
脱氧胞苷产量的效果更优,为2
′‑
脱氧胞苷的生产提供了新的思路与方法。
[0038]
(2)采用本发明所述的重组微生物生产2
′‑
脱氧胞苷,其产量更高,且发酵周期短,生产强度高,可用于2
′‑
脱氧胞苷的大规模工业化生产。
[0039]
(3)本发明所提供的重组微生物的构建方法是一种定向的理性的菌种构建方法,相比传统诱变方法更为高效便捷,可操作性强。
附图说明
[0040]
图1为2
′‑
脱氧胞苷代谢路径示意图。
[0041]
其中,pyre:乳清酸磷酸核糖转移酶;nrda、nrdb:核糖核苷二磷酸还原酶;nrdc:硫氧还蛋白;yfbr:dcmp磷酸水解酶;yfdr:5
′‑
脱氧核苷酸酶;dcd:dctp脱氨酶;cdd:胞苷/脱氧胞苷脱氨酶;tdk:胸苷/脱氧尿苷激酶;udk:尿苷/胞苷激酶;dut:脱氧核苷酸三磷酸酶;thya:胸苷酸合成酶;deoa:胸苷磷酸化酶;omp:乳清酸-5
′‑
单磷酸;ump:尿嘧啶核糖核苷酸;udp:尿苷二磷酸;utp:尿苷三磷酸;cr:胞苷;cytosine:胞嘧啶;cmp:胞苷一磷酸;cdp:胞苷二磷酸;ctp:胞苷三磷酸;dudp:脱氧尿苷二磷酸;dutp:脱氧尿苷三磷酸;dcdp:脱氧胞苷二磷酸;dctp:脱氧胞苷三磷酸;dcmp:脱氧胞苷一磷酸;dcr:2
′‑
脱氧胞苷;dump:脱氧尿苷一磷酸;dur:脱氧尿苷;dtmp:脱氧胸苷一磷酸;dtdp:脱氧胸苷二磷酸;dttp:脱氧胸苷三磷酸。
[0042]
图2为产物和副产物相应hplc出峰图。
[0043]
图3为核糖核苷二磷酸还原酶nrdba及硫氧还蛋白nrdc过表达对2
′‑
脱氧胞苷产量影响检测结果图。
[0044]
图4为yfdr对2
′‑
脱氧胞苷产量影响检测结果图。
[0045]
图5为yfdr和yfbr共表达对2
′‑
脱氧胞苷产量影响检测结果图。
[0046]
图6为yfdr、yfbr和nrdbca共表达对2
′‑
脱氧胞苷产量影响检测结果图。
具体实施方式
[0047]
下面结合具体实施例,对本发明作进一步详细的阐述,下述实施例不用于限制本发明,仅用于说明本发明。以下实施例中所使用的实验方法如无特殊说明,实施例中未注明具体条件的实验方法,通常按照常规条件,下述实施例中所使用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0048]
本发明的技术方案可以应用大肠杆菌,枯草杆菌,谷氨酸棒杆菌,乳杆菌或者其他的微生物,下面以大肠杆菌为例,对技术方案进行进一步说明。
[0049]
实验方法1:基因敲除方法
[0050]
本发明采用datsenko的方法进行微生物基因敲除,基于red重组系统,用具有50-nt同源延伸的引物通过pcr生成的可选择的抗生素耐药基因替换目的基因序列,从而达到基因敲除的目的。本发明所采用的具体基因敲除的方法记载于文献:k.a.datsenko,b.l.wanner.one-step inactivation of chromosomal genes in escherichia coli k-12using pcr products.proceedings of the national academy sciences of the usa,2000,97(12):6640-6645。相应的基因敲除引物见baba2006.mol syst biol,2(1)0008。
[0051]
实验方法2:摇瓶发酵验证重组菌株生产2
′‑
脱氧胞苷的方法
[0052]
1、试剂
[0053]
(1)发酵培养基:每升培养基中葡萄糖30g,5n-5倍的盐溶液200ml,tm3溶液1ml,柠檬酸铁10mg,七水硫酸镁246mg,氯化钙111mg,硫胺素1μg,以无菌去离子水定容至1l。
[0054]
其中5n-5倍的盐溶液为十二水合磷酸氢二钠75.6g,磷酸二氢钾每升15g,氯化钠2.5g,氯化铵25g,以离子水定容至1l;tm3溶液为四水氯化锌2.0g,六水氯化钙2.0g,两水钼酸钠2.0g,五水硫酸铜1.9g,硼酸0.5g,盐酸100ml,去离子水定容至1l。
[0055]
上述溶液进行高压蒸汽灭菌,灭菌温度121℃,时间20-30min。
[0056]
(2)lb培养基:每升培养基中含酵母粉5g,氯化钠10g,蛋白胨10g,去离子水定容至1l(著[美]j.莎姆布鲁克.黄培堂译,分子克隆指南2002,1595)。
[0057]
上述溶液进行高压蒸汽灭菌,灭菌温度121℃,时间20-30min。
[0058]
2、仪器:恒温摇床培养箱。
[0059]
3、方法:
[0060]
摇瓶发酵过程如下:(1)接种重组菌株至含有抗生素的4ml的lb培养基中,置37℃摇床250rpm培养;(2)取培养16h后的种子500μl转移至2ml含有抗生素的lb液体培养基,于37℃摇床250rpm培养4h;(3)将2.5ml二级种子全部转入装有18ml发酵培养基的摇瓶中,置37℃摇床中,250rpm培养4h;(4)添加iptg至终浓度1mm后,调节摇床温度至34℃,继续培养20h左右,取1ml发酵液离心后(12000rpm,1min),取上清检测,检测方法详见实验方法3。
[0061]
实验方法3:hplc测定发酵液中的2
′‑
脱氧胞苷
[0062]
吸取发酵液用水稀释至一定的倍数,离心过0.22μm的滤膜,用高效液相色谱(hplc)检测。hplc的参数如下:采用xbridgec18 4.6*150mm5um,流动相为甲醇和10mm乙酸铵或pbs(ph 4.0),流动相比例0.01-4.5分钟甲醇比例为2%上升到20%,4.5-4.6分钟甲醇比例由20%下降到2%,4.6-8分钟甲醇比例维持2%,利用紫外检测器检测波长260nm;初始流动相的流速为1.0ml/min,发酵液的上样量2μl,柱温30℃。2
′‑
脱氧胞苷出峰时间为6.55分钟,乳清酸出峰时间为2.48分钟,胞苷出峰时间为4.16分钟,hplc图谱如图2所示。
[0063]
实施例1:构建不能利用和降解2
′‑
脱氧胞苷的重组大肠杆菌
[0064]
如图1所示,在大肠杆菌中2
′‑
脱氧胞苷的流向有两个:第一条路径为2
′‑
脱氧胞苷进入dump回补途径,经胞苷/脱氧胞苷脱氨酶(cdd)催化,将生成的2
′‑
脱氧胞苷转变成2
′‑
脱氧尿苷(dur),dur在脱氧尿苷激酶(tdk)催化下生成dump,并进一步通过胸苷酸合成酶(thya)催化生成dtmp,dtmp再生成dtdp和dttp等,作为细胞dna合成的前体;第二条路径是2
′‑
脱氧胞苷被胸苷磷酸化酶(deoa)催化,降解为胞嘧啶。因此,为了使细胞中更多地积累2
′‑
脱氧胞苷,需要敲除cdd、deoa等基因,截断补救和降解路径。
[0065]
本发明以大肠杆菌w3110(atcc27325)为出发菌株,按照实验方法1在w3110中敲除利用和降解2
′‑
脱氧胞苷的基因cdd、deoa,构建菌株sha57(w3110δcddδdeoa)(表1)。
[0066]
表1.菌株改造
[0067]
[0068][0069]
大肠杆菌有三条已知的有氧从头合成dump(dtmp合成的底物)的途径:dctp、dutp和2
′‑
脱氧胞苷路径。大肠杆菌中的dump有75-80%是由dctp路径合成(neuhard,j.,and r.kelln.1996.biosynthesis and conversions of pyrimidines,p.580
–
599.in f.c.neidhardt,r.curtiss iii,j.l.ingraham,e.c.c.lin,k.b.low,b.magasanik,w.s.reznikoff,m.riley,m.schaechter,and h.e.umbarger(ed.),escherichia coli and salmonella:cellular and molecular biology,2nd ed.asm press,washington,dc.)。dctp路径的第一步是dcd基因编码的dctp脱氨酶将dctp转化为dutp,然后dutp二磷酸酶(dut)催化dutp的焦磷酸盐水解,产生dump,剩余的dump合成主要是通过dudp路径进行的,2
′‑
脱氧胞苷路径则是其补救路径。2
′‑
脱氧胞苷是由dcmp水解得到,而dcmp是dctp二磷酸酶催化水解得到,为了增加2
′‑
脱氧胞苷的产量则需要增加前体dcmp和dctp的含量,因此需要截断dctp流向dutp,使代谢集中流向2
′‑
脱氧胞苷路径。因此在菌株sha57(w3110δcddδdeoa)基础上敲除dcd,构建得到sha60(w3110δcddδdeoaδdcd)(表1)。
[0070]
大肠杆菌w3110摇瓶发酵检测不到2
′‑
脱氧胞苷,菌株sha60摇瓶发酵可以检测到极少量的2
′‑
脱氧胞苷4.7mg/l左右,还含有大量乳清酸(~240mg/l)。因为在大肠杆菌k-12菌中,乳清酸磷酸核糖转移酶(pyre)上游rph基因的c端缺失一个碱基造成移码突变,翻译提前终止,导致pyre的表达不足,使得在嘧啶合成过程中会出现乳清酸的积累,因此通过在质粒或基因组上对乳清酸磷酸核糖转移酶(pyre)基因进行超表达来解除上游rph基因移码突变导致的表达缺陷,减少乳清酸的积累。核苷/h+协同转运蛋白(nupg)会将胞外的核苷转运到胞内,可以敲除nupg阻止2
′‑
脱氧胞苷进入细胞,进而积累更多的2
′‑
脱氧胞苷,因此选择敲除nupg同时整合ptrc-pyre-kanfrt,最后构建得到菌株sha65(w3110δcddδdeoaδdcdδnupg::ptrc-pyre),发酵检测无乳清酸积累。
[0071]
sha65菌株整合ptrc-pyre的具体构建过程如下:
[0072]
首先,构建整合基础质粒pez07-pyre-kanfrt:分别以w3110基因组和pkd4为模板,用下表2引物pyre-f/pyre-r、k-f/k-r分别扩增含pyre和kanfrt片段,得到大小分别为680bp、1499bp的片段且电泳无杂带,直接进行过柱回收纯化(捷瑞胶回收纯化试剂盒),得到的片段以纳摩尔比1:2与ncoi/xhoi酶切回收的pez07载体片段进行ez克隆构建(苏州神洲基因gbclonart无缝克隆试剂盒),重组克隆反应液于45℃水浴锅温浴30min,然后转移到冰上放置5min,转入tg1化转感受态细胞,42℃热击2min,冰浴2min后加入800μl复苏培养基lb,复苏培养1h后离心涂布含100mg/l壮观霉素和50mg/l卡那霉素抗性的lb平板,次日挑取克隆培养过夜,提取质粒进行酶切验证,最终构建得到质粒pez07-pyre-kanfrt。
[0073]
然后,以pez07-pyre-kanfrt为模板,用表2中引物nupg-kinf/nupg-kinr扩增敲除整合片段,得到大小为2856bp的无杂带的片段,直接进行过柱回收纯化,取约500ng的片段加入到sha60/pkd46的电转化感受态细胞,混匀,转入0.1cm电击杯中,用电击仪作电转化。
电击条件:200ω,25μf,电击电压1.8kv,电击时间为5ms,电击后迅速加入800μl的lb,37℃,200rpm复苏培养1h,之后涂于含50ng/ml卡那霉素的lb平板,37℃过夜培养。次日,将电转化得到的转化子以nupg-200f/kan-yzr进行菌液pcr验证,得到大小为2043bp片段,确认sha65k构建成功,随后转化pcp20进行消抗,最终得到sha65菌株。
[0074]
随后在sha65菌株基础上敲除大肠杆菌胞苷/尿苷激酶编码基因udk,构建得到sha85k(w3110δcddδdeoaδdcdδnupg::ptrc-pyreδudk::kanfrt),并转化pcp20完成消抗,最终得到菌株sha85。sha85和对照菌株sha65摇瓶发酵dcr产量分别为6.5mg/l和14.78mg/l,dcr产量提高2倍左右,说明udk敲除是有效果的,我们通过体外酶催化实验也验证了这一结果。
[0075]
大肠杆菌胞苷-尿苷激酶udk,主要功能是以atp或gtp作为磷酸供体,催化胞苷或尿苷生成5
′‑
单磷酸胞苷或5
′‑
单磷酸尿苷;此外大肠杆菌还有胸苷/2
′‑
脱氧尿苷激酶tdk,主要是催化胸苷或2
′‑
脱氧尿苷生成5
′‑
单磷酸胸苷或5
′‑
单磷酸-2
′‑
脱氧尿苷,但是并没有关于催化2
′‑
脱氧胞苷生成相应5
′‑
单磷酸-2
′‑
脱氧胞苷的相关研究和报道。本实验室在bl21(de3)菌株中进行体外过表达获得tdk和udk的粗酶液,进行体外酶催化实验,以1g/l 2
′‑
脱氧胞苷和3g/latp作为底物,37℃水浴反应1h检测5
′‑
单磷酸-2
′‑
脱氧胞苷(dcmp的生成),同时设计水替代酶切为阴性对照检测底物添加量是否准确,bl21(de3)/pet28a酶液为不表达对照,结果显示:用水代替酶液的组分只能检测到2
′‑
脱氧胞苷950mg/l,和我们加入的含量接近;添加对照bl21(de3)/pet28a酶液的样品既没有2
′‑
脱氧胞苷,也没有dcmp检测到,可能是菌株cdd没有敲除,2
′‑
脱氧胞苷全部转化为2
′‑
脱氧尿苷;而只有添加udk酶液的样品能检测到356mg/l的dcmp及165mg/l 2
′‑
脱氧胞苷,其他的2
′‑
脱氧胞苷可能通过cdd转化为2
′‑
脱氧尿苷。这一结果显示udk不仅可以催化胞苷、尿苷生成相应的核苷单磷酸,也可以催化2
′‑
脱氧胞苷生成dcmp,这是关于udk具有此功能的首次研究和报道,为2
′‑
脱氧胞苷的菌株改造提供基础。
[0076]
表2.菌株sha65构建和验证引物
[0077][0078]
实施例2:核糖核苷二磷酸还原酶及硫氧还蛋白编码基因过表达可以显著提高2
′‑
脱氧胞苷产量
[0079]
大肠杆菌中2
′‑
脱氧胞苷主要是作为dump合成的补救路径:2
′‑
脱氧胞苷在特异性的dcmp磷酸水解酶(yfbr)催化下生成2
′‑
脱氧尿苷(dur),dur在胸苷/脱氧尿苷尿苷激酶
(tdk)催化下生成dump,并进一步通过胸苷酸合成酶(thya)催化生成dtmp,dtmp再生成dtdp和dttp等,作为细胞dna合成的前体(weiss b.the deoxycytidine pathway for thymidylate synthesis in escherichia coli.j bacteriol.2007;189:7922
–
7926.doi:10.1128/jb.00461-07.)。因此为了积累更多2
′‑
脱氧胞苷,需要将特异性的dcmp磷酸水解酶(yfbr)进行过表达,构建质粒pez07-yfbr,命名px109。sha57转化px109的重组菌株摇瓶发酵后2
′‑
脱氧胞苷产量为15.7mg/l,说明yfbr过表达确实有助于加快dcmp到2
′‑
脱氧胞苷水解,提高2
′‑
脱氧胞苷产量。且dcd基因敲除菌株sha60转化px109后的重组菌株摇瓶发酵后2
′‑
脱氧胞苷产量分别提高到43.8mg/l,说明阻断dctp到dutp路径有利于积累更多的2
′‑
脱氧胞苷。
[0080]
专利us6777209和us7387893中公开了在产胸苷的大肠杆菌宿主中表达t4噬菌体来源的核糖二磷酸还原酶(nrda、nrdb)和硫氧还蛋白(nrdc)通过提高dudp和dcdp直接或间接提高dutp的含量从而提高胸苷产量,而且kim 2015在进行2
′‑
脱氧胞苷菌株改造也过表达nrdcba,主要是拉动cdp快速转变成dcdp,进而磷酸化成dctp,最终到2
′‑
脱氧胞苷的前体dcmp。因此本专利在pez07-yfbr的基础上过表达nrdbca,构建表达质粒pez07-yfbr-nrdbca,命名pha289。
[0081]
共表达质粒pha289分别转化基因工程菌sha60、sha65和sha85发酵比较dcr产量和其他副产物情况。结果如图3所示:对照菌株sha60/px109的2
′‑
脱氧胞苷产量为55.36mg/l,但是有150mg/l的乳清酸积累;sha60/pha289过表达nrdbca后dcr产量提高4倍左右,说明核糖核苷二磷酸还原酶及硫氧还蛋白nrdbca过表达可以显著提高dcr产量,但是副产物乳清酸也明显积累,达到1.2g/l,因此在此菌株的基础上整合ptrc-pyre解决乳清酸积累,构建得到sha65,sha65/pha289发酵dcr产量变化不大,但是乳清酸基本没有了;随后的udk敲除菌株sha85转化pha289后dcr产量也明显提高到370mg/l。
[0082]
目前结果显示nrdbca过表达可以明显提高dcr产量,说明整个代谢过程中nrdbca的过表达比较重要,可以有效的拉动主路径的cdp到dcdp,间接提高2
′‑
脱氧胞苷的前体dcmp含量,提高2
′‑
脱氧胞苷的产量。为了便于后续工作的开展,因此先将ptrc-nrdbca整合1-2个拷贝到基因工程菌sha85上拓宽2
′‑
脱氧胞苷前体合成的代谢流,再考虑过表达dcmp水解催化生成2
′‑
脱氧胞苷。按照实施例1中ptrc-pyre整合到基因组构建过程,先构建基础整合质粒pez07-nrdbca-kanfrt,命名pt12k,然后以此质粒为模板,选择pepa和fima位点,分别设计50bp同源臂的敲除整合引物,扩增得到敲除整合片段,先构建一个拷贝ptrc-nrdbca整合菌株sha92,然后在sha92基础上整合第二个拷贝ptrc-nrdbca得到整合2个拷贝的整合菌株sha95。
[0083]
下面以px109质粒为例介绍表达质粒的构建过程:
[0084]
以w3110基因组为模板,用下表4引物yfbr-f/yfbr-r扩增得到大小为642bp的yfbr片段,电泳无杂带直接进行过柱回收纯化(捷瑞胶回收纯化试剂盒),得到的片段与ncoi/xhoi酶切回收的pez07载体片段进行ez克隆构建(苏州神洲基因gbclonart无缝克隆试剂盒),重组克隆反应液于45℃水浴锅温浴30min,然后转移到冰上放置5min,转入tg1化转感受态细胞,42℃热击2min,冰浴2min后加入800μl复苏培养基lb,复苏培养1h后离心涂布含100mg/l壮观霉素抗性的lb平板,次日挑取克隆用引物pcl-f/pcl-r为模板扩增验证,大小为1100bp的对应克隆接种壮观霉素抗性培养基培养过夜,提取质粒进行酶切验证,最终构
建得到质粒pez07-yfbr,命名px109,再在此基础上连接nrdbca,构建共表达质粒pez07-yfbr-nrdbca,命名pha289。
[0085]
表3.质粒信息表
[0086]
编号质粒信息pez07psc101ori,aada1px109pez07-yfbrpha289pez07-yfbr-nrdbcapt12kfpez07-nrdbca-kanfrtpha294pez07-yfdrpha302pez07-yfdr-nrdbcapha340pez07-yfbr-yfdrpha346pez07-yfdr-yfbr
[0087]
表4.px109质粒构建引物
[0088]
引物序列5
′‑3′
yfbr-fgattaaataaggaggaataaaccatgaaacagagccatttttttgccyfbr-rggtaccagctgcagatctcgagttacagcggtgaatcctggcpcl-fgacatcataacggttctggcpcl-raaaacagccaagctggagac
[0089]
实施例3:5
′‑
脱氧核苷酸酶yfdr可以更高效的催化dcr生产
[0090]
大肠杆菌中yfbr基因编码dcmp磷酸水解酶(dcmp phosphohydrolase,ec3.1.3.89),yfbr主要是催化胞内不同的脱氧核苷单核苷酸如damp、dtmp、dimp、dump、dcmp和dgmp等生成对应的2
′‑
脱氧核苷。zimmerman等人在2008年对大肠杆菌中的yfbr的底物特异性和催化机制展开研究(zimmerman md,proudfoot m,yakunin a,minor w.structural insight into the mechanism of substrate specificity and catalytic activity of an hd-domain phosphohydrolase:the 5
′‑
deoxyribonucleotidase yfbr from escherichia coli.j mol biol.2008apr 18;378(1):215-26.doi:10.1016/j.jmb.2008.02.036.epub 2008mar 4.pmid:18353368;pmcid:pmc2504004.),文中揭示已经鉴定的具有脱氧核苷水解酶活性的除了yfbr还有sure、yjjg,此外还有yedj和yfdr等。zimmerman等人在2008年研究比较显示,yfdr和yfbr类似也是一类含有hd结构域的蛋白,可催化各类脱氧单核酸生成相应的2
′‑
脱氧核苷,yfdr的底物范围比yfbr更广,底物亲和力更低,yfdr尤其是对底物dgmp和damp有很好的催化速率。虽然zimmerman等在2008年揭示比较了多种5
′‑
核苷酸酶,但是kim et al(2015)构建的重组微生物发酵法还是通过过表达dcmp磷酸水解酶编码基因yfbr进行dcr生产。而本专利在2
′‑
脱氧核苷生产过程中筛选比较了各种5
′‑
核苷酸酶,如yfbr、sure、yjjg、yedj、usha、cdh和yfdr等,从中发现了yfdr对2
′‑
脱氧胞苷的产量有很好的促进作用,其次是yfbr,其他5
′‑
核苷酸酶过表的效果不明显。
[0091]
构建yfbr和yfdr过表达质粒px109和pha294(按照上述实施例2中质粒px109质粒为例设计构建表达质粒pez07-yfdr)分别转化菌株sha65和sha85,分别得到重组菌株sha65/px109、sha65/pha294、sha85/px109和sha85/pha294等,根据实验方法2进行摇瓶发
酵,结果如图4所示:yfdr过表达菌株的dcr产量大约是yfbr过表达菌株的3倍左右,无论是sha65还是sha85菌株,yfdr基因过表达可以催化dcr生成,且催化效率明显高于yfbr基因过表达,说明yfdr可以更高效的催化dcr的生产,这是关于利用超表达yfdr过表达生产dcr的首次报道。此外两种重组菌株都有极少量副产物,2mg/l左右的尿嘧啶和15mg/l左右的副产物胸腺嘧啶。
[0092]
实施例4:yfdr和yfbr共表达可明显提高dcr产量
[0093]
如上述实施3的结果显示yfdr基因过表达可以在改造菌株中实现dcr生产,且dcr产量明显高于yfbr基因过表达菌株,因此在此基础上将两个基因进行共表达构建,考虑到基因表达是线性的,距离启动子越近的基因可能表达更好,因此分别构建质粒pez07-yfbr-yfdr和pez07-yfdr-yfbr比较是否有差别,分别命名pha340和pha346。
[0094]
将共表达质粒pha340和pha346连同单基因表达质粒px109和pha294分别转化到重组菌株sha65进行摇瓶发酵初筛,得到的重组菌株按照实验方法2进行摇瓶发酵比较。结果如图5所示:yfdr和yfbr比较结果和实施例3结果一致,yfdr单表达菌株dcr产量明显比yfbr单表达菌株产量高,副产物只有及少量的尿嘧啶和胸腺嘧啶。将yfdr和yfbr按照不同的顺序进行串联表达后dcr产量都明显提高,且明显是1+1》2的效果,dcr产量高达565.78mg/l,这也是首次报道将yfdr和yfbr共同表达来进行dcr的生产。yfdr和yfbr的不同表达顺序dcr产量差别不大,但是副产物差别较大,pha340(即pez07-yfbr-yfdr)过表达菌株的副产物偏少,但是pha346(即pez07-yfdr-yfbr)过表达菌株的tdr和dur两种副产物明显增加,说明基因不同顺序表达存在区别。
[0095]
随后将这些质粒转化整合一个拷贝ptrc-nrdbca的基因工程菌sha92,进一步验证yfdr和yfbr串联表达的联用效果。将共表达质粒pha340和pha34和单基因表达质粒px109和pha294分别转化sha92菌株感受态,得到重组菌株按照实验方法2进行摇瓶发酵比较。发酵结果如图6,和上述sha65转化菌株的结果比较一致,且在整合一个拷贝ptrc-nrdbca后过表达yfdr和yfbr,dcr产量高达845.97mg/l,yfdr和yfbr的不同表达顺序可以看出副产物明显不同,副产物胞苷含量差不多都是15.67mg/l,差异较大的副产物是胸苷tdr和脱氧尿苷dur,pha340(即pez07-yfbr-yfdr)过表达菌株的两种副产物都是偏少的,分别是77.54mg/l和39.58mg/l,但是pha346(即pez07-yfdr-yfbr)过表达菌株的两种副产物分别高达141.97mg/l和133.61mg/l,可能是由于yfdr的催化底物更广泛,因此相对来说pha346(即pez07-yfdr-yfbr)最终得到的副产物量更高。综合考虑目的产物dcr含量和副产物的含量,选择pha340作为最优的表达质粒。
[0096]
最后,在整合两个拷贝ptrc-nrdbca的菌株sha95中过表达pha340质粒,副产物的种类和含量与整合一个拷贝的sha92菌株中过表达pha340差不多,但是dcr产量明显提高到992mg/l,可以达到工业化生产dcr的水平。
[0097]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1