一种1,4-二氮杂环烷类化合物的制备方法
1.本发明涉及有机合成技术领域,尤其是涉及一种1,4-二氮杂环烷类化合物的制备方法。
背景技术:
2.目前,1,4-二氮杂环烷类化合物分为二氮六元杂环化合物和二氮七元杂环化合物,其中二氮六元杂环化合物在医药、化工、表面活性剂、催化剂等领域有发挥重要作用。在药物化学领域,以哌嗪及其衍生物为基础的药物合成利福平、氟哌酸、吡哌酸、哌嗪硫酸盐、哌嗪磷酸盐等医药用品,是治疗肿瘤、感染、精神疾病、血液系统等疾病的重要内容。然而,大多数重氮杂环,尤其是含哌嗪的药物,通常具有n取代,缺乏c取代多样性。而二氮七元杂环化合物已有多种经典的反应能够得到氮杂七元环化合物,其中环加成反应可有效构建氮杂七元环及其衍生物。然而,二氮七元杂环化合物的合成非常有限,另一个局限性是缺乏c取代的多样性。
3.因此,开发一种高效简单的方法来快速构建具有含四元碳中心的的1,4-二氮杂环烷类化合物尤为重要。
技术实现要素:
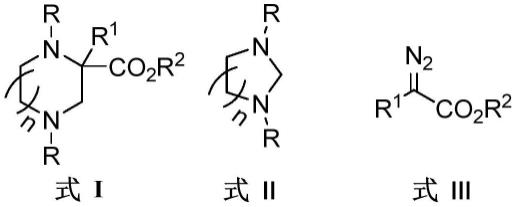
4.本发明旨在至少解决现有技术中存在的技术问题之一。为此,本发明的第一方面提出1,4-二氮杂环烷类化合物。
5.根据本发明的第一方面实施例的1,4-二氮杂环烷类化合物,包括如下步骤:
6.将式ii所示的化合物与式iii所示的化合物在铜催化剂和有机溶剂存在下进行反应,得到式i所示结构的化合物:
[0007][0008]
其中,n为1、2或3;
[0009]
r选自取代或未取代的苯基、苄基;
[0010]
r1选自h、c
2~4
的酯基、五元杂环基、取代或未取代的苯基;
[0011]
r2选自c
1~10
的烷基、苄基;
[0012]
r和r1中任选含有的取代基选自卤素、c
1~6
的烷基。
[0013]
根据本发明实施例的1,4-二氮杂环烷类化合物的制备方法,至少具有如下有益效果:
[0014]
本发明由式ii所示的化合物与式iii所示的化合物在铜催化剂的作用下,经环加
成制备含四元碳中心的1,4-二氮杂环烷类化合物,该合成方法的原料廉价易得、操作方便、产率高,为含四元碳中心的1,4-二氮杂环烷类化合物提供了一种简洁高效的制备方法。
[0015]
根据本发明的一些实施例,r选自取代或未取代的苯基、苄基;
[0016]
r1选自h、甲酸甲酯基、噻吩基、取代或未取代的苯基;
[0017]
r2选自c
1~10
的烷基、苄基;
[0018]
r和r1中任选含有的取代基选自氟、溴、c
1~3
的烷基。
[0019]
根据本发明的一些实施例,所述铜催化剂选自溴化亚铜、溴化铜、氯化铜、三氟甲磺酸铜、三氟甲磺酸亚铜、三苯基磷溴化亚铜或六氟磷酸四(乙腈)铜中的至少一种。
[0020]
根据本发明的一些实施例,所述铜催化剂选自三氟甲磺酸铜、溴化亚铜、六氟磷酸四(乙腈)铜中的至少一种。由此,当铜催化剂选自三氟甲磺酸铜、溴化亚铜、六氟磷酸四(乙腈)铜中的至少一种时,能够进一步提升本发明的产率。
[0021]
根据本发明的一些实施例,所述有机溶剂选自二氯甲烷、1,2-二氯乙烷、甲苯、氯苯、n,n-二甲基甲酰胺、乙腈或四氢呋喃中的至少一种。
[0022]
根据本发明的一些实施例,所述式ii所示的化合物、式iii所示的化合物和铜催化剂的摩尔比为1:1~5:0.05~0.5。
[0023]
根据本发明的一些实施例,所述反应的温度为0℃~100℃。由此,当反应的温度为0℃~100℃时,能够促进反应的进行。
[0024]
根据本发明的一些实施例,所述反应的温度为60℃~100℃。由此,能够进一步提升本发明的产率。
[0025]
根据本发明的一些实施例,所述反应的时间为2h~48h。
[0026]
根据本发明的一些实施例,所述反应的时间为2h~3h。
[0027]
定义和一般术语
[0028]“c
1~10
的烷基”表示碳原子总数为1-10的烷基,包括c
1-10
的直链烷基、c
1-10
的支链烷基和c
3-10
的环烷基,例如可以为碳原子总数为1、2、3、4、5、6、7、8、9或10的直链烷基、碳原子总数为1、2、3、4、5、6、7、8、9或10的支链烷基或者碳原子总数为3、4、5、6、7、8、9或10的环烷基,例如可以为甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、异戊基、正己基、环丙基、甲基环丙基、乙基环丙基、环戊基、甲基环戊基、环己基等。针对“c
1-6
的烷基”和“c
1-3
的烷基”具有与此相似的解释,所不同的是,碳原子数不同。
[0029]“卤素”表示为氟、氯、溴、碘中的任意一个或两个以上。
[0030]“c
2~4
的酯基”表示为碳原子总数为2~4的酯基,其结构式为-coor3。
[0031]“五元杂环基”表示为被n、s、o中的一个或多个取代的五元环烷基,例如是噻吩基、呋喃基或吡咯基。
[0032]“取代或未取代的苯基”表示苯环上任选一个h基被本文所定义的基团取代。
[0033]
本发明的其它特征和优点将在随后的说明书中阐述,并且,部分地从说明书中变得显而易见,或者通过实施本发明而了解。
附图说明
[0034]
本发明的上述和/或附加的方面和优点从结合下面附图对实施例的描述中将变得明显和容易理解,其中:
[0035]
图1为本发明实施例1制得的产物1a的1h nmr谱图;
[0036]
图2为本发明实施例1制得的产物1a的
13
c nmr谱图;
[0037]
图3为本发明实施例2制得的产物1b的1h nmr谱图;
[0038]
图4为本发明实施例2制得的产物1b的
13
c nmr谱图;
[0039]
图5为本发明实施例3制得的产物1c的1h nmr谱图;
[0040]
图6为本发明实施例3制得的产物1c的
13
c nmr谱图;
[0041]
图7为本发明实施例4制得的产物1d的1h nmr谱图;
[0042]
图8为本发明实施例4制得的产物1d的
13
c nmr谱图;
[0043]
图9为本发明实施例5制得的产物1e的1h nmr谱图;
[0044]
图10为本发明实施例5制得的产物1e的
13
c nmr谱图;
[0045]
图11为本发明实施例6制得的产物1f的1h nmr谱图;
[0046]
图12为本发明实施例6制得的产物1f的
13
c nmr谱图;
[0047]
图13为本发明实施例1制得的产物1g的1h nmr谱图;
[0048]
图14为本发明实施例1制得的产物1g的
13
c nmr谱图;
[0049]
图15为本发明实施例2制得的产物1h的1h nmr谱图;
[0050]
图16为本发明实施例2制得的产物1h的
13
c nmr谱图;
[0051]
图17为本发明实施例3制得的产物1i的1h nmr谱图;
[0052]
图18为本发明实施例3制得的产物1i的
13
c nmr谱图;
[0053]
图19为本发明实施例4制得的产物1j的1h nmr谱图;
[0054]
图20为本发明实施例4制得的产物1j的
13
c nmr谱图;
[0055]
图21为本发明实施例5制得的产物1k的1h nmr谱图;
[0056]
图22为本发明实施例5制得的产物1k的
13
c nmr谱图;
[0057]
图23为本发明实施例6制得的产物1l的1h nmr谱图;
[0058]
图24为本发明实施例6制得的产物1l的
13
c nmr谱图;
[0059]
图25为本发明对比例1制得的产物的1h nmr谱图。
具体实施方式
[0060]
以下是本发明的具体实施例,并结合实施例对本发明的技术方案作进一步的描述,但本发明并不限于这些实施例。
[0061]
本发明所采用的试剂、方法和设备,如无特殊说明,均为本技术领域常规试剂、方法和设备。
[0062]
实施例和对比例用到的原料如下:
[0063]
所有铜催化剂:购自安耐吉化学;
[0064]
金催化剂:(2-二叔丁基膦基-2',4',6'-三异丙基-1,1'-联苯基)氯化金,购自安耐吉化学;
[0065]
铑催化剂:醋酸铑,购自乐研试剂;
[0066]
银催化剂:硝酸银,购自安耐吉化学;
[0067]
铁催化剂:氯化铁,购自安耐吉化学;
[0068]
化合物2a、2g、2h、2i和3a~3k的制备方法如下:
[0069]
化合物2a:
[0070][0071]
在装有20mln,n-二甲基甲酰胺的反应瓶中加入1,3-二溴丙烷(10mmol),随后在室温下加入对甲苯胺i和碳酸钾并回流24h,通过tlc监测,直至反应完成。反应完后用乙酸乙酯稀释,用饱和碳酸氢钠洗涤有机相,分离有机层,用无水硫酸钠干燥,真空下旋干,得到粗产物ii。在装有20ml甲醇的反应瓶中加入水合醛,0.5ml乙酸。30℃下反应,通过tlc监测直至反应完成;经过滤干燥得到白色固体化合物2a。
[0072]
化合物2g-2i:
[0073][0074]
在装有40ml异丙醇的反应瓶中加入取代胺iii(10mmol),转移冰浴中,0℃下加入乙二醛(5mmol)并在0℃下反应,通过tlc监测,直至反应完成。经过滤干燥或萃取得到iv。在装有40ml四氢呋喃的反应瓶中加入iv,转移冰浴中,0℃下缓慢加入硼氢化钠。30℃下反应12h,反应完后用水淬灭,用乙酸乙酯萃取三次,用水和饱和食盐水洗涤有机相。分离有机层,用无水硫酸钠干燥,真空下旋干,得到粗产物v。在装有20ml甲醇的反应瓶中加入v、水合醛和0.5ml乙酸,30℃下反应,通过tlc监测直至反应完成。经过滤干燥得到2g和2h,通过柱层析纯化得到2i。
[0075]
化合物3a-3k:
[0076][0077]
在装有40ml乙腈的反应瓶中加入vi(6mmol)和对乙酰氨基苯磺酰叠氮(6.6mmol),0℃下加入1,8-二偶氮杂双螺环[5.4.0]十一-7-烯(9mmol),30℃下反应12h。待反应完加入水和二氯甲烷,用饱和食盐水洗涤,分离有机层,经无水硫酸钠干燥。真空下旋干,经柱层析法纯化得到化合物3a~3k。
[0078]
实施例1
[0079]
本实施例提供1,4-二氮杂环烷类化合物(1a)的制备方法,步骤如下:
[0080][0081]
在25ml反应瓶中氮气保护下,化合物3a(264mg,1.5mmol)溶于二氯甲烷(5.0ml)中,将上述溶液利用注射泵缓慢注射入化合物2a(266mg,1.0mmol)和三氟甲磺酸铜(36mg,0.1mmol)的二氯甲烷(5.0ml)溶液中,反应体系在80℃下搅拌3小时,除去溶剂后粗产品利用硅胶柱层析分离得到白色固体产物1a(307mg,产率:74%)。
[0082]
如图1和图2,所得产品1a的检测数据如下:1h nmr(500mhz,cdcl3):δ7.62-7.54(m,2h),7.33-7.27(m,2h),7.26-7.22(m,1h),6.94(d,j=8.3hz,2h),6.83(d,j=8.4hz,2h),6.58(d,j=8.6hz,2h),6.47(d,j=8.6hz,2h),4.48(d,j=15.8hz,1h),4.03-3.96(m,1h),3.86-3.76(m,1h),3.69-3.58(m,2h),3.25-3.14(m,4h),2.31-2.22(m,1h),2.19(s,3h),2.16(s,3h),2.09-2.01(m,1h);
13
c nmr(125mhz,cdcl3):δ174.1,148.5,147.7,138.7,129.7,128.9,128.5,128.3,127.4,127.3,126.9,116.3,114.6,73.5,64.4,52.2,52.1,49.5,30.9,20.40,20.36.
[0083]
实施例2
[0084]
本实施例提供1,4-二氮杂环烷类化合物(1b)的制备方法,步骤如下:
[0085][0086]
在25ml反应瓶中氮气保护下,化合物3b(327mg,1.5mmol)溶于二氯甲烷(5.0ml)中,将上述溶液利用注射泵缓慢注射入化合物2a(266mg,1.0mmol)和三氟甲磺酸铜(36mg,0.1mmol)的二氯甲烷(5.0ml)溶液中,反应体系在80℃下搅拌3小时,除去溶剂后粗产品利用硅胶柱层析分离得到白色固体产物1b(307mg,产率:73%)。
[0087]
如图3和图4,所得产品1b的检测数据如下:1h nmr(500mhz,cdcl3):δ7.69-7.62(m,2h),7.31-7.26(m,2h),7.24-7.19(m,1h),6.93(d,j=8.4hz,2h),6.83(d,j=8.4hz,2h),6.62(d,j=8.6hz,2h),6.53(d,j=8.6hz,2h),4.34(d,j=15.7hz,1h),4.02-3.95(m,1h),3.86-3.78(m,1h),3.61(d,j=15.7hz,1h),3.58-3.51(m,1h),3.22-3.14(m,1h),2.27-2.18(m,4h),2.16(s,3h),2.09-2.01(m,1h),1.06(s,9h);
13
c nmr(125mhz,cdcl3):δ172.1,149.5,147.9,139.7,129.6,128.8,128.7,128.1,127.7,127.1,126.9,117.1,115.6,81.9,74.5,64.8,52.2,49.1,30.5,27.5,20.4,20.4.
[0088]
实施例3
[0089]
本实施例提供1,4-二氮杂环烷类化合物(1c)的制备方法,步骤如下:
[0090][0091]
在25ml反应瓶中氮气保护下,化合物3c(291mg,1.5mmol)溶于二氯甲烷(5.0ml)中,将上述溶液利用注射泵缓慢注射入化合物2a(266mg,1.0mmol)和三氟甲磺酸铜(36mg,0.1mmol)的二氯甲烷(5.0ml)溶液中,反应体系在80℃下搅拌3小时,除去溶剂后粗产品利用硅胶柱层析分离得到白色固体产物1c(324mg,产率:75%)。
[0092]
如图5和图6,所得产品1c的检测数据如下:1h nmr(500mhz,cdcl3):δ7.65-7.54(m,2h),7.01-6.92(m,4h),6.86(d,j=8.3hz,2h),6.56(d,j=8.4hz,2h),6.47(d,j=8.4hz,2h),4.39(d,j=15.8hz,1h),4.02(dd,j=15.2,6.7hz,1h),3.83(dd,j=15.9,8.6hz,1h),3.69-3.57(m,2h),3.35-3.20(m,4h),2.32-2.24(m,1h),2.21(s,3h),2.18(s,3h),2.13-2.05(m,1h);
13
c nmr(125mhz,cdcl3):δ174.1,162.1(d,j=243.8hz),148.5,147.5,134.5(d,j=2.5hz),130.4(d,j=8.8hz),129.7,129.0,127.6,127.2,116.4,115.0(d,j=21.3hz),114.7,73.164.7,52.3,52.2,49.5,31.0,20.38,20.35.
[0093]
实施例4
[0094]
本实施例提供1,4-二氮杂环烷类化合物(1d)的制备方法,步骤如下:
[0095][0096]
在25ml反应瓶中氮气保护下,化合物3d(171mg,1.5mmol)溶于二氯甲烷(5.0ml)中,将上述溶液利用注射泵缓慢注射入化合物2a(266mg,1.0mmol)和三氟甲磺酸铜(36mg,0.1mmol)的二氯甲烷(5.0ml)溶液中,反应体系在80℃下搅拌3小时,除去溶剂后粗产品利用硅胶柱层析分离得到无色油状产物1d(226mg,产率:64%)。
[0097]
如图7和图8,所得产品1d的检测数据如下:1h nmr(500mhz,cdcl3):δ6.93(dd,j=12.8,8.4hz,4h),6.64(d,j=8.6hz,2h),6.47(d,j=8.6hz,2h),4.56(dd,j=10.8,5.7hz,1h),4.38(dd,j=15.7,5.7hz,1h),4.29-4.23(m,1h),4.23-4.16(m,1h),3.97(d,j=14.6hz,1h),3.75(dd,j=7.6,2.1hz,2h),3.59(dd,j=15.6,10.8hz,1h),3.10(m,1h),2.19-2.03(m,7h),1.76-1.70(m,1h),1.30(t,j=7.1hz,3h);
13
c nmr(125mhz,cdcl3):δ174.0,146.9,144.3,130.2,129.8,126.04,126.00,112.0,61.2,58.6,52.7,51.1,46.7,28.0,20.3,20.3,14.5.
[0098]
实施例5
[0099]
本实施例提供1,4-二氮杂环烷类化合物(1e)的制备方法,步骤如下:
[0100][0101]
在25ml反应瓶中氮气保护下,化合物3a(237mg,1.5mmol)溶于二氯甲烷(5.0ml)中,将上述溶液利用注射泵缓慢注射入化合物2e(266mg,1.0mmol)和三氟甲磺酸铜(36mg,0.1mmol)的二氯甲烷(5.0ml)溶液中,反应体系在80℃下搅拌3小时,除去溶剂后粗产品利用硅胶柱层析分离得到无色油状产物1e(285mg,产率:72%)。
[0102]
如图9和图10,所得产品1e的检测数据如下:1h nmr(500mhz,cdcl3):δ6.98(d,j=8.4hz,2h),6.98(d,j=8.4hz,2h),6.85(d,j=8.5hz,2h),6.79(d,j=8.6hz,2h),4.27(s,2h),3.77-3.72(m,2h),3.47(s,6h),3.38(t,j=6.0hz,2h),2.23(s,3h),2.22(s,3h),2.13-2.07(m,2h);
13
c nmr(125mhz,cdcl3):δ170.1,147.9,130.5,129.6,129.2,127.6,120.4,114.9,76.0,58.0,52.5,51.6,51.0,29.8,20.6,20.4.
[0103]
实施例6
[0104]
本实施例提供1,4-二氮杂环烷类化合物(1f)的制备方法,步骤如下:
[0105][0106]
在25ml反应瓶中氮气保护下,化合物3f(374mg,1.5mmol)溶于二氯甲烷(5.0ml)中,将上述溶液利用注射泵缓慢注射入化合物2a(266mg,1.0mmol)和三氟甲磺酸铜(36mg,0.1mmol)的二氯甲烷(5.0ml)溶液中,反应体系在80℃下搅拌3小时,除去溶剂后粗产品利用硅胶柱层析分离得到红色油状产物1f(317mg,产率:65%)。
[0107]
如图11和图12,所得产品1f的检测数据如下:1h nmr(500mhz,cdcl3):δ7.34(d,j=7.4hz,1h),7.23-7.17(m,3h),7.09(t,j=7.7hz,1h),7.00-6.95(m,2h),6.86-6.79(m,3h),6.74(d,j=8.3hz,2h),6.63(d,j=7.8hz,1h),6.57(d,j=8.3hz,2h),6.27(d,j=8.5hz,2h),5.08(d,j=15.6hz,1h),4.62(d,j=15.7hz,1h),4.23-4.15(m,1h),4.03(d,j=15.7hz,1h),3.83-3.76(m,1h),3.76-3.70(m,1h),3.59(d,j=15.7hz,1h),3.24-3.16(m,1h),2.35-2.21(m,2h),2.16(s,6h);
13
c nmr(125mhz,cdcl3):δ177.7,149.7,147.9,141.3,135.7,131.4,131.0,129.4,129.1,128.7,128.4,127.7,127.5,127.4,126.6,122.74,122.68,115.1,109.4,70.6,61.9,51.5,51.1,43.7,32.4,20.7,20.3.
[0108]
实施例7
[0109]
本实施例提供哌嗪类化合物(1g)的制备方法,步骤如下:
[0110][0111]
在25ml反应瓶中氮气保护下,化合物3g(264mg,1.5mmol)溶于二氯甲烷(5.0ml)中,将上述溶液利用注射泵缓慢注射入化合物2g(252mg,1.0mmol)和三氟甲磺酸铜(36mg,0.1mmol)的二氯甲烷(5.0ml)溶液中,反应体系在80℃下搅拌3小时,除去溶剂后粗产品利用硅胶柱层析分离得到白色固体产物1g(356mg,产率:90%)。
[0112]
如图13和图14,所得产品1g的检测数据如下:1h nmr(500mhz,cdcl3):δδ7.32(dd,j=7.7,2.0hz,2h),7.24-7.16(m,3h),7.06(d,j=8.4hz,2h),6.87(d,j=8.5hz,2h),6.78(d,j=8.4hz,2h),6.70(d,j=8.5hz,2h),4.26(dd,j=11.8,2.1hz,1h),3.90-3.80(m,4h),3.71-3.66(m,1h),3.54-3.48(m,1h),3.19(d,j=11.8hz,1h),3.14(td,j=11.4,3.7hz,1h),2.26(s,3h),2.15(s,3h);
13
c nmr(125mhz,cdcl3):δ172.7,149.1,146.8,138.9,130.7,129.8,129.7,128.30,128.28,127.88,127.86,124.2,117.0,72.3,64.0,52.1,50.4,49.8,20.7,20.6.
[0113]
实施例8
[0114]
本实施例提供哌嗪类化合物(1h)的制备方法,步骤如下:
[0115][0116]
在25ml反应瓶中氮气保护下,重氮化合物3h(285mg,1.5mmol)溶于二氯甲烷(5.0ml)中,将上述溶液利用注射泵缓慢注射入咪唑烷化合物2g(252mg,1.0mmol)和三氟甲磺酸铜(36mg,0.1mmol)的二氯甲烷(5.0ml)溶液中,反应体系在80℃下搅拌3小时,除去溶剂后粗产品利用硅胶柱层析分离得到白色固体产物1h(377mg,产率:91%)。
[0117]
如图15和图16,所得产品1h的检测数据如下:1h nmr(500mhz,cdcl3):δ7.19(d,j=8.3hz,2h),7.05(d,j=8.2hz,2h),7.00(d,j=8.0hz,2h),6.85(d,j=8.5hz,2h),6.78(d,j=8.3hz,2h),6.71(d,j=8.6hz,2h),4.23(dd,j=11.8,2.2hz,1h),3.86
–
3.81(m,4h),3.69
–
3.63(m,1h),3.52
–
3.46(m,1h),3.17(d,j=11.8hz,1h),3.11(td,j=11.3,3.7hz,1h),2.25(s,6h),2.15(s,3h);
13
c nmr(125mhz,cdcl3):δ172.8,149.1,146.8,137.5,135.9,130.6,129.7,129.0,128.3,127.7,124.2,116.9,72.0,63.9,52.0,50.3,49.7,21.1,20.7,20.6.
[0118]
实施例9
[0119]
本实施例提供哌嗪类化合物(1i)的制备方法,步骤如下:
[0120][0121]
在25ml反应瓶中氮气保护下,重氮化合物3i(273mg,1.5mmol)溶于二氯甲烷(5.0ml)中,将上述溶液利用注射泵缓慢注射入咪唑烷化合物2g(252mg,1.0mmol)和三氟甲磺酸铜(36mg,0.1mmol)的二氯甲烷(5.0ml)溶液中,反应体系在80℃下搅拌3小时,除去溶剂后粗产品利用硅胶柱层析分离得到白色固体产物1i(377mg,产率:52%)。
[0122]
如图17和图18,所得产品1i的检测数据如下:1h nmr(500mhz,cdcl3):δ7.15(dd,j=2.8,1.1hz,1h),7.12(dd,j=5.0,3.0hz,1h),7.07(d,j=8.3hz,2h),6.91(dd,j=5.0,1.0hz,1h),6.86(d,j=8.5hz,2h),6.83(d,j=8.4hz,2h),6.69(d,j=8.5hz,2h),4.24(dd,j=11.7,2.0hz,1h),3.86(td,j=12.0,3.5hz,1h),3.81(s,3h),3.63
–
3.59(m,1h),3.54
–
3.46(m,1h),3.20(d,j=11.7hz,1h),3.11(td,j=11.3,3.6hz,1h),2.27(s,3h),2.18(s,3h);
13
c nmr(125mhz,cdcl3):δ172.8,148.9,146.9,140.0,131.0,129.9,129.8,128.4,127.8,125.1,123.8,123.3,117.1,69.1,63.3,52.1,49.9,49.8,20.7,20.6.
[0123]
实施例10
[0124]
本实施例提供哌嗪类化合物(1j)的制备方法,步骤如下:
[0125][0126]
在25ml反应瓶中氮气保护下,重氮化合物3j(2 37mg,1.5mmol)溶于二氯甲烷(5.0ml)中,将上述溶液利用注射泵缓慢注射入咪唑烷化合物2g(252mg,1.0mmol)和三氟甲磺酸铜(36mg,0.1mmol)的二氯甲烷(5.0ml)溶液中,反应体系在80℃下搅拌3小时,除去溶剂后粗产品利用硅胶柱层析分离得到无色油状产物1j(115mg,产率:30%)。
[0127]
如图19和图20,所得产品1j的检测数据如下:1h nmr(500mhz,cdcl3):δ7.11
–
7.06(m,4h),7.04(d,j=8.5hz,2h),6.86(d,j=8.4hz,2h),3.81(s,2h),3.68(s,6h),3.60(t,j=5.3hz,2h),3.26(t,j=5.3hz,2h),2.28(s,3h),2.27(s,3h);
13
c nmr(125mhz,cdcl3):δ169.2,148.9,147.0,133.0,130.3,129.8,129.2,123.6,117.4,73.6,59.2,52.7,49.7,48.8,20.9,20.6.
[0128]
实施例11
[0129]
本实施例提供哌嗪类化合物(1k)的制备方法,步骤如下:
[0130][0131]
在25ml反应瓶中氮气保护下,重氮化合物3k(264mg,1.5mmol)溶于二氯甲烷(5.0ml)中,将上述溶液利用注射泵缓慢注射入咪唑烷化合物2h(382mg,1.0mmol)和三氟甲磺酸铜(36mg,0.1mmol)的二氯甲烷(5.0ml)溶液中,反应体系在80℃下搅拌3小时,除去溶剂后粗产品利用硅胶柱层析分离得到白色固体产物1k(410mg,产率:77%)。
[0132]
如图21和图22,所得产品1k的检测数据如下:1h nmr(500mhz,cdcl3):δ7.34-7.26(m,4h),7.25-7.20(m,3h),7.06(d,j=8.8hz,2h),6.78(d,j=8.8hz,2h),6.65(d,j=8.8hz,2h),4.26(d,j=12.0hz,1h),3.84(s,3h),3.79(td,j=12.6,3.3hz,1h),3.72(dt,j=12.6,3.4hz,1h),3.52(d,j=10.7hz,1h),3.22(d,j=12.1hz,1h),3.16(td,j=11.1,3.7hz,1h);
13
c nmr(125mhz,cdcl3):δ172.2,149.7,148.1,138.0,132.0,130.6,128.6,128.3,127.6,125.2,118.0,114.0,112.4,72.3,62.7,52.3,49.8,48.9.
[0133]
实施例12
[0134]
本实施例提供哌嗪类化合物(1l)的制备方法,步骤如下:
[0135][0136]
在25ml反应瓶中氮气保护下,重氮化合物3g(264mg,1.5mmol)溶于二氯甲烷(5.0ml)中,将上述溶液利用注射泵缓慢注射入咪唑烷化合物2i(252mg,1.0mmol)和三氟甲磺酸铜(36mg,0.1mmol)的二氯甲烷(5.0ml)溶液中,反应体系在80℃下搅拌3小时,除去溶剂后粗产品利用硅胶柱层析分离得到白色固体产物1l(340mg,产率:77%)。
[0137]
如图23和图24,所得产品1l的检测数据如下:1h nmr(500mhz,cdcl3):δ7.49-7.36(m,2h),7.33-7.19(m,12h),7.19-7.13(m,1h),3.88(d,j=15.1hz,1h),3.85(s,3h),3.70(d,j=15.1hz,1h),3.62(d,j=13.3hz,1h),3.40-3.31(m,2h),2.92(td,j=11.8,3.2hz,1h),2.82-2.77(m,1h),2.77-2.71(m,1h),2.42-2.33(m,2h);
13
c nmr(125mhz,cdcl3):δ173.7,140.9,140.2,138.2,129.1,128.4,128.2,128.09,128.07,127.97,127.7,127.1,126.5,73.2,64.1,62.8,55.0,54.2,51.2,46.8.
[0138]
实施例13~18
[0139]
实施例13~18提供一系列1,4-二氮杂环烷类化合物的制备方法,其原料和制备方法同实施例7,其区别在于,铜催化剂不相同,具体如表1。
[0140]
表1
[0141] 铜催化剂产率%实施例13氯化铜56实施例14溴化亚铜66实施例15三氟甲磺酸亚铜45实施例16四氟硼酸四乙氰铜61实施例17六氟磷酸四(乙腈)铜64实施例18乙酰丙酮酸铜49
[0142]
实施例19~23
[0143]
实施例19~23提供一系列1,4-二氮杂环烷类化合物的制备方法,其原料和制备方法同实施例7,其区别在于,反应温度不相同,具体如表2。
[0144]
表2
[0145] 反应温度/℃产率%实施例19032实施例203072实施例214075实施例226078实施例2310089
[0146]
实施例24~29
[0147]
实施例24~29提供一系列1,4-二氮杂环烷类化合物的制备方法,其原料和制备方法同实施例7,其区别在于,有机溶剂不相同,具体如表3。
[0148]
表3
[0149] 有机溶剂产率%实施例241,2-二氯乙烷68实施例25甲苯32实施例26氯苯43实施例27六氟异丙醇47实施例28乙腈48实施例29四氢呋喃54
[0150]
对比例1~4
[0151]
对比例1~4提供一系列1,4-二氮杂环烷类化合物的制备方法,其原料和制备方法同实施例7,其区别在于,催化剂不相同,具体如表4。
[0152]
表4
[0153][0154]
对比例1虽然能够采用铑催化剂进行反应,但是得到的产物是混合物,无法分离出目标产物,如图25所示,图25是核磁共振氢谱图,从图上可以看出有许多未知峰型,得到的是一种混合物,无法分离得到单一产物。
[0155]
上面结合本发明实施例作了详细说明,但本发明不限于上述实施例,在所属技术领域普通技术人员所具备的知识范围内,还可以在不脱离本发明宗旨的前提下作出各种变化。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1