2-氯-嘧啶-5-甲醛的制备方法与流程
1.本发明涉及有机合成领域,具体而言,涉及一种2-氯-嘧啶-5-甲醛的制备方法。
背景技术:
2.嘧啶类化合物,一种在六元环的1位和3位上有两个氮原子的芳香族化学分子,因其具有优异的生物活性而受到了广泛的研究关注。嘧啶类化合物具有多种生物学特性,包括抗结核、抗生素、抗真菌、抗病毒、抗炎、抗疟疾、抗癌和抗肿瘤作用以及抗hiv活性。其中,2-氯-嘧啶-5-甲醛是一个重要的药物中间体,且目前国内外对高效、低成本、环境友好的2-氯嘧啶-5-甲醛中间体制备工艺的要求越来越高。
3.关于2-氯-嘧啶-5-甲醛的合成技术,目前关于研究合成的路线很少,现有技术中如cn109369539a公开了一种2-氯嘧啶-5-甲醛的合成方法报道:使用格氏法合成2-氯嘧啶-5-碳醛,其合成路线如下所示:
[0004][0005]
但是,该制备方法原料成本高且性质不稳定,具有高风险因素,这不仅提高了制造成本,也增加了生产风险,并不适宜大规模工业化应用。
[0006]
现有技术中如cn109928933a也公开了一种2-氯嘧啶-5-甲醛及其制备方法报道:用盐酸氯甲脒处理阿诺德盐,其合成路线如下所示:
[0007][0008]
但是,该方法存在一些安全问题,如吸湿性强,对原料具有腐蚀性,以及制造成本高及收率低。
[0009]
综上,现有技术中的2-氯嘧啶-5-甲醛制备方法均存在原料昂贵,难以获得,反应风险高以及环保性较差等问题,故而,有必要提供一种新的2-氯嘧啶-5-甲醛制备方法,以改善上述问题。
技术实现要素:
[0010]
本发明的主要目的在于提供一种2-氯-嘧啶-5-甲醛的制备方法,以解决现有技术中在制备2-氯-嘧啶-5-甲醛时存在的成本高、风险高、环保性较差等问题。
[0011]
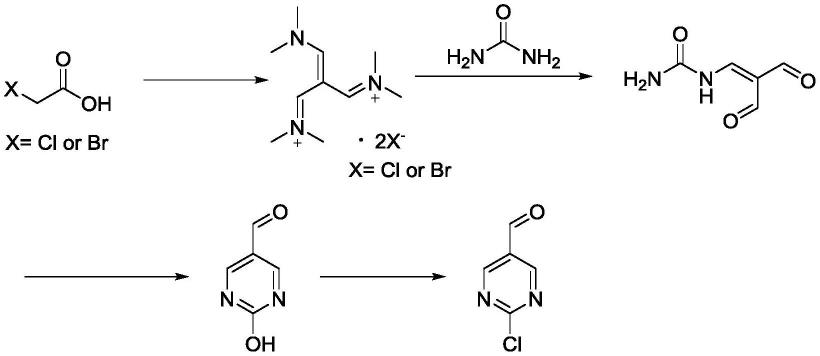
为了实现上述目的,根据本发明的一个方面,提供了一种2-氯-嘧啶-5-甲醛的制备方法,制备方法包括以下步骤:步骤s1:对乙酸类化合物进行甲酰化反应,得到中间产物a;步骤s2:使包含有中间产物a和尿素的溶液进行缩合反应,得到中间产物b;步骤s3:对中间产物b进行闭环反应,得到中间产物c;步骤s4:对中间产物c进行氯化反应,得到2-氯-嘧
醛基嘧啶而言,具有原料成本更低廉的优点。且各反应过程中涉及到的中间产物均稳定性更佳,例如本发明上述过程中的中间产物a即为一种稳定性非常好的阿诺德盐,便于长期储存和运输。同时,本发明避免了在制备过程中使用具有强腐蚀性的氯甲脒盐酸盐,环保性更佳。尤其是,上述反应过程操作更简单、流程更简单、反应安全性更高。从而,基于上述制备过程,本发明原料成本更低、利用率更高、中间产物稳定性更佳、反应过程更平稳、且产物收率更高,进而更适用于工业化批量生产,工业化应用前景更佳。
附图说明
[0022]
构成本技术的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
[0023]
图1示出了本发明实施例1中制备得到的中间化合物a的核磁氢谱图;
[0024]
图2示出了本发明实施例1中制备得到的中间化合物b的核磁氢谱图;
[0025]
图3示出了本发明实施例1中制备得到的中间化合物c的核磁氢谱图;
[0026]
图4示出了本发明实施例1中制备得到的2-氯嘧啶-5-甲醛的核磁氢谱图;
[0027]
图5示出了本发明实施例1中制备得到的2-氯嘧啶-5-甲醛的气相色谱-质谱图。
具体实施方式
[0028]
需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互组合。下面将参考附图并结合实施例来详细说明本发明。
[0029]
正如本发明背景技术部分所描述的,现有技术中在制备2-氯-嘧啶-5-甲醛时存在成本高、风险高、产物收率低等问题。为了解决这一问题,本发明提供了一种2-氯-嘧啶-5-甲醛的制备方法,制备方法包括以下步骤:步骤s1:对乙酸类化合物进行甲酰化反应,得到中间产物a;步骤s2:使包含有中间产物a和尿素的溶液进行缩合反应,得到中间产物b;步骤s3:对中间产物b进行闭环反应,得到中间产物c;步骤s4:对中间产物c进行氯化反应,得到2-氯-嘧啶-5-甲醛;其中,乙酸类化合物为中间产物a为x表示cl或br;中间产物b为中间产物c为
[0030]
本发明上述制备方法的合成路线如下所示:
[0031][0032]
基于此,本发明以特定的乙酸类化合物作为反应原料,相较于现有技术中的2-甲氧基-5-醛基嘧啶而言,具有原料成本更低廉的优点。且各反应过程中涉及到的中间产物均稳定性更佳,例如本发明上述过程中的中间产物a即为一种稳定性非常好的阿诺德盐,便于长期储存和运输。同时,本发明避免了在制备过程中使用具有强腐蚀性的氯甲脒盐酸盐,环保性更佳。尤其是,上述反应过程操作更简单、流程更简单、反应安全性更高。从而,基于上述制备过程,本发明原料成本更低、利用率更高、中间产物稳定性更佳、反应过程更平稳、且产物收率更高,进而更适用于工业化批量生产,工业化应用前景更佳。
[0033]
在一种优选的实施方式中,步骤s1包括:将乙酸类化合物、n-n-二甲基甲酰胺及三氯氧磷混合以进行甲酰化反应。优选地,乙酸类化合物、n-n-二甲基甲酰胺及三氯氧磷的摩尔比为1:(12~14):(8~9)。基于此,甲酰化反应的反应过程更平稳、操作过程更安全,且乙酸类化合物的利用率更高,中间产物a的产率更高。
[0034]
为了进一步提高甲酰化反应的效率,从而进一步提高产物收率,在一种优选的实施方式中,乙酸类化合物为溴乙酸时,步骤s1包括:在惰性气体氛围下,在0~5℃温度条件下,先向n-n-二甲基甲酰胺中以0.05~0.1ml/s的滴速滴加三氯氧磷,再向n-n-二甲基甲酰胺中每隔5~15min、分3~8次加入溴乙酸,形成第一混合液;使第一混合液在85~95℃温度条件下搅拌5~10h后,再在0~5℃温度条件下,向第一混合液中以0.05~0.1ml/s的滴速依次滴加质量浓度为35~45%的溴化氢水溶液及四氢呋喃,形成第二混合液;使第二混合液在20~25℃温度条件下搅拌20~40min,得到中间产物a。优选地,溴化氢水溶液的用量为溴乙酸重量的1.0~1.2倍;四氢呋喃的用量为溴乙酸重量的8~12倍。在上述溴化氢水溶液及四氢呋喃的滴加过程中,有固体物质析出,在氮气保护下对反应后料液进行过滤,滤饼依次用乙酸和乙腈淋洗。将淋洗后固体于1l单口瓶中60℃减压干燥1h后再于真空干燥箱60℃中减压干燥6h,即得到中间产物a
[0035]
为了进一步提高甲酰化反应的效率,从而进一步提高产物收率,在一种优选的实施方式中,乙酸类化合物为氯乙酸时,步骤s1包括:在惰性气体氛围下,在0~5℃温度条件下,先向n-n-二甲基甲酰胺中加入氯乙酸,再向n-n-二甲基甲酰胺中以0.05~0.1ml/s的滴
速滴加三氯氧磷,形成第四混合液;使第四混合液依次在75~85℃温度条件下搅拌0.5~1.5h、90~100℃温度条件下搅拌0.5~1.5h、100~110℃温度条件下搅拌0.5~1.5h后,再在20~25℃温度条件下,向第四混合液中以0.05~0.1ml/s的滴速依次滴加质量浓度为30~36%的浓盐酸及四氢呋喃搅拌20~40min,得到中间产物a。优选地,浓盐酸的用量为氯乙酸重量的0.8~1.2倍;四氢呋喃的用量为氯乙酸重量的8~12倍。在上述浓盐酸及四氢呋喃的滴加过程中,有固体析出。对反应后料液进行过滤,滤饼用thf淋洗后于真空干燥箱中60℃真空干燥6h,即得到中间产物a
[0036][0037]
在一种优选的实施方式中,步骤s2包括:将中间产物a、尿素及水混合以进行缩合反应;优选地,中间产物a和尿素的摩尔比为1:(1.9~2.2)。基于此,缩合反应的反应过程更平稳、操作过程更安全,且中间产物b的产率更高。为了进一步平衡缩合反应的效率及稳定性,在一种优选的实施方式中,缩合反应的反应温度为20~25℃,反应时间为20~30h。缩合反应结束后,反应液由黄色变为透明,对反应后料液进行过滤,滤饼用水淋洗后在鼓风干燥箱中50℃干燥6h,得到白色粉末,即为中间产物b。
[0038]
在一种优选的实施方式中,步骤s3包括:将中间产物b溶解于乙酸中后,先在105~115℃温度条件下回流1~3h,再去除体系中乙酸,然后向体系中加入甲苯继续回流0.5~2h,得到中间产物c。基于此,闭环反应的反应过程更平稳、操作过程更安全,且中间产物c的产率更高。为了进一步平衡缩合反应的效率及稳定性,在一种优选的实施方式中,中间产物b、乙酸和甲苯的摩尔比为1:(20~30):(10~15)。加入甲苯继续回流0.5~2h后,将料液降温至室温并对其进行过滤,滤饼用甲苯淋洗,即得到中间产物c。
[0039]
为了进一步提高产物收率,在一种优选的实施方式中,步骤s4包括:将中间产物c溶解于乙腈后,在0~5℃温度条件下,向体系中以0.02~0.05ml/s的滴速依次滴加n,n-二异丙基乙胺及三氯氧磷形成第五混合液;使第五混合液在75~85℃温度条件下搅拌4~8h后,再在20~25℃温度条件下调控体系ph值为6.5~7.5,得到2-氯-嘧啶-5-甲醛。其中,可通过tlc监控反应进程,待tlc显示无原料剩余后,将反应后料液降温至室温,滴加碳酸钠水溶液调节ph至上述范围,再加乙酸乙酯进行萃取,萃取后的有机相采用水洗涤后旋干。将旋干后粗品加入至mtbe:正庚烷=1:2的料液中搅拌30min,再对料液进行过滤,得到的固体即为2-氯-嘧啶-5-甲醛。优选地,中间产物c、n,n-二异丙基乙胺及三氯氧磷的摩尔比为1:(1.1~1.2):(1.4~1.6)。
[0040]
以下结合具体实施例对本技术作进一步详细描述,这些实施例不能理解为限制本技术所要求保护的范围。
[0041]
实施例1
[0042]
(1)中间产物a的制备
[0043]
于2l圆底四口瓶中加入dmf(315.6g,4.32mol),氮气置换三次。冰水浴降温至0℃。于氮气保护下控温0~5℃滴加pocl3(441.3g,2.88mol),约30min滴毕。溴乙酸(50g,
0.36mol)分5次在氮气保护下加入反应,每次间隔10分钟。
[0044]
反应移入油浴锅升温至90℃,氮气保护下搅拌6h。需注意酸性尾气吸收。反应由无色透明变为红色液体。反应降温至0℃,于氮气保护下滴加40%hbr(58.2g,0.72mol)后滴加thf(500ml)。反应于室温搅拌30min,有固体析出。在氮气保护下过滤,滤饼依次用乙酸(360ml)和乙腈(250ml)淋洗。固体于1l单口瓶中60℃减压干燥1h后于真空干燥箱60℃中减压干燥6h。得到白色吸湿固体73.2g,收率48%。
[0045]
hnmr检测如图1所示,结果如下:
[0046]1h-nmr(400mhz,[d6]-dmso):δ=8.82(s,3h,ch),3.58(s,9h,ch3),3.47(s,9h,ch3)ppm.
[0047]
(2)中间产物b的制备
[0048]
于250ml三口瓶中加入中间产物a(10g,36.77mmol)。将尿素(4.41g,73.47mmol)溶于20ml水后加入反应瓶。加入70ml水后在室温下搅拌24h。
[0049]
反应液由黄色变为透明,由白色固体洗出。过滤,滤饼用20ml水淋洗后在鼓风干燥箱中50℃干燥6h。得到白色粉末4.3g,收率82.4%。
[0050]
hnmr检测如图2所示,结果如下:
[0051]1h-nmr(400mhz,[d6]-dmso):δ=11.26(d,j=3.3hz;1h,nh),9.84(d,j=3.4hz;1h,cho),9.53(s,1h,cho),8.28(dd,j=12.9hz,j=3.2hz;1h,ch),7.83(s,1h,nh2),7.53(s,1h,nh2)ppm.
[0052]
(3)中间产物c的制备:
[0053]
在100ml圆底烧瓶中加入中间产物b(4.5g,31.7mmol),加入50ml冰乙酸溶解,于110℃回流2h,得到浅棕色粗品。旋干冰乙酸,加入50ml甲苯于110℃回流1h反应降温至室温,过滤,滤饼用10ml甲苯淋洗,得到浅黄色固体3.8g,收率96.7%。
[0054]
hnmr检测如图3所示,结果如下:
[0055]1h-nmr(400mhz,[d6]-dmso):δ=12.67(s,1h,oh),9.65(s,2h,ch),8.81(s,1h,cho)ppm.
[0056]
(4)2-氯-嘧啶-5-甲醛的制备:
[0057]
于250ml三口瓶中加入中间产物c(5g,40.29mmol)。加50ml乙腈溶解。反应降温至0℃,滴加n,n-二异丙基乙胺(6.25g,48.35mmol)及三氯氧磷(9.27g,60.44mmol)后,反应升温至80℃,搅拌6h。tlc显示无原料剩余。反应降温至室温,滴加碳酸钠水溶液调节ph至中性,加乙酸乙酯20ml萃取。有机相加20ml水洗涤后旋干得到棕黄色粗品。粗品加mtbe:正庚烷=1:2后搅拌30min,过滤,得到4.8g浅黄色固体,收率83.5%。
[0058]
hnmr检测如图4所示,结果如下:
[0059]1h-nmr(400mhz,cdcl3):δ=10.2(s,1h,cho),9.2(s,2h,ch)ppm.
[0060]
图5示出了本发明实施例1中制备得到的2-氯嘧啶-5-甲醛的气相色谱-质谱图。
[0061]
实施例2
[0062]
(1)中间产物a的制备
[0063]
将2-氯乙酸(10.00g,0.11mol)溶解于dmf(92.82g,1.27mmol),加入1l圆底四口瓶,氮气置换3次。反应冰水浴降温至0℃,滴加pocl3(129.8g,0.85mol)。滴毕,反应升温至80℃搅拌1h。继续升温至95℃搅拌1h。继续升温至105℃搅拌1h。反应降温至室温,滴加浓盐
酸(7.72g,0.22mol)后滴加thf(200ml),搅拌30min。有固体析出。过滤,滤饼用thf(40ml)淋洗后于真空干燥箱中60℃真空干燥6h。得到白色固体21.10g,收率68.8%。
[0064]
hnmr检测结果如下:
[0065]1h-nmr(400mhz,d2o):δ=8.30(s,3h,ch),3.57(s,9h,ch3),3.40(s,9h,ch3)ppm.
[0066]
(2)中间产物b的制备
[0067]
于250ml三口瓶中加入中间产物a(10g,36.77mmol)。将尿素(4.41g,73.47mmol)溶于20ml水后加入反应瓶。加入70ml水后在室温下搅拌24h。
[0068]
反应液由黄色变为透明,由白色固体洗出。过滤,滤饼用20ml水淋洗后在鼓风干燥箱中50℃干燥6h。得到白色粉末4.4g,收率84.3%。
[0069]
1h-nmr(400mhz,[d6]-dmso):=11.26(d,j=3.3hz;1h,nh),9.84(d,j=3.4hz;1h,cho),9.53(s,1h,cho),8.28(dd,j=12.9hz,j=3.2hz;1h,ch),7.83(s,1h,nh2),7.53(s,1h,nh2)ppm.
[0070]
(3)中间产物c的制备:
[0071]
在100ml圆底烧瓶中加入中间产物b(4.5g,31.7mmol),加入50ml冰乙酸溶解,于110℃回流2h,得到浅棕色粗品。旋干冰乙酸,加入50ml甲苯于110℃回流1h反应降温至室温,过滤,滤饼用10ml甲苯淋洗,得到浅黄色固体3.9g,收率99.1%。
[0072]
1h-nmr(400mhz,[d6]-dmso):=12.67(s,1h,oh),9.65(s,2h,ch),8.81(s,1h,cho)ppm.
[0073]
(4)2-氯-嘧啶-5-甲醛的制备:
[0074]
于250ml三口瓶中加入中间产物c(5g,40.29mmol)。加50ml乙腈溶解。反应降温至0℃,滴加n,n-二异丙基乙胺(6.25g,48.35mmol)及三氯氧磷(9.27g,60.44mmol)后,反应升温至80℃,搅拌6h。tlc显示无原料剩余。反应降温至室温,滴加碳酸钠水溶液调节ph至中性,加乙酸乙酯20ml萃取。有机相加20ml水洗涤后旋干得到棕黄色粗品。粗品加mtbe:正庚烷=1:2后搅拌30min,过滤,得到4.7g浅黄色固体,收率81.7%。
[0075]1h-nmr(400mhz,cdcl3):δ=10.2(s,1h,cho),9.2(s,2h,ch)ppm.
[0076]
以上仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1