哌嗪苯基氨基取代的嘧啶氨基酸衍生物及制备方法与应用
1.本发明属于有机化合物合成及医药应用领域,涉及哌嗪苯基氨基取代的嘧啶氨基酸衍生物及制备方法与应用。
背景技术:
2.在恶性肿瘤中常发现组蛋白去乙酰化酶(hdac)异常表达,而采用hdac抑制剂(hdaci)可以抑制hdac活性,使得组蛋白乙酰化水平提高,激活某些特定基因表达,从而抑制癌细胞增值、诱导肿瘤细胞凋亡,故hdac被认为是治疗血液恶性肿瘤的的有效靶点(参见blood2016;127(18):2168-2170和blood2019;(supplement_1):4325)。fms样酪氨酸激酶3(fms-like receptor tyrosine kinase,flt3)是一种跨膜蛋白,在急性髓性白血病(aml)的白血病原始细胞上均匀表达,且对血液系统恶性肿瘤和自体免疫疾病的的生物学和临床管理具有重要意义(参见blood2018;131(15):1631-1632和blood2019;134(supplement_1):1441)。临床前及临床研究表明,hdac抑制剂和flt3抑制剂显示出有效的协调的抗白血病作用(参见:blood2018;132(supplement_1):1427),因此设计合成结构新颖、具有成药性的新型hdac/flt3双靶点药物,对于治疗血液系统恶性肿瘤及自体免疫疾病来说具有十分重要的意义。
技术实现要素:
3.本发明的目的在于提供一类具有hdac/flt3双重抑制活性且具有抗肿瘤作用的哌嗪苯基氨基取代的嘧啶氨基酸衍生物及其制备方法与应用,该类化合物生物活性优良,同时提升了化合物的分子多样性和新颖性。
4.为了实现上述目的,本公开的技术方案为:
5.在本发明的第一方面,本发明提供了一种哌嗪苯基氨基取代的嘧啶氨基酸衍生物或其药学上可接受的盐,哌嗪苯基氨基取代的嘧啶氨基酸衍生物为通式i所示的哌嗪苯基氨基取代的嘧啶氨基酸衍生物i或通式ii所示的哌嗪苯基氨基取代的嘧啶氨基酸衍生物ii:
[0006][0007]
其中,n为1,2,3,4,5,6,7。
[0008]
优选地,n为2,3,4,5,6。
[0009]
3-(5-氟-2-((4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)氨基)丙酸甲酯(i-1)
[0010]
4-(5-氟-2-((4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)氨基)丁酸甲酯(i-2)
[0011]
5-(5-氟-2-((4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)氨基)戊酸甲酯(i-3)
[0012]
6-(5-氟-2-((4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)氨基)己酸甲酯(i-4)
[0013]
7-(5-氟-2-((4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)氨基)庚酸甲酯(i-5)
[0014]
3-((5-氟-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)氨基)-n-羟基丙酰胺(ii-1)
[0015]
4-((5-氟-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)氨基)-n-羟基丁酰胺(ii-2)
[0016]
5-((5-氟-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)氨基)-n-羟基戊酰胺(ii-3)
[0017]
6-((5-氟-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)氨基)-n-羟基己酰胺(ii-4)
[0018]
7-((5-氟-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)氨基)-n-羟基庚酰胺(ii-5)
[0019]
上述10个化合物名称后的括号中为其相应的代号,为叙述方便和表达简洁,上述括号中的代号在本说明书以下内容中将被直接应用。
[0020]
本发明所述的化合物可以游离形式或进一步以盐的形式存在,目的是为了提高水溶性,增加生物利用度。
[0021]
本发明所述的“药学上可接受的盐”是指常规的无毒性的盐,主要包括本技术化合物的碱性氨基所形成的盐。这些盐是本领域熟练技术人员熟知的,熟练的技术人员可以制备本领域知识所提供的任何药学上可接受的盐。此外,熟练技术人员还可以根据溶解度、稳定性、容易制剂等因素取某种盐而舍去另一种盐。这些盐的测定和最优化在熟练技术人员的经验范围内。
[0022]
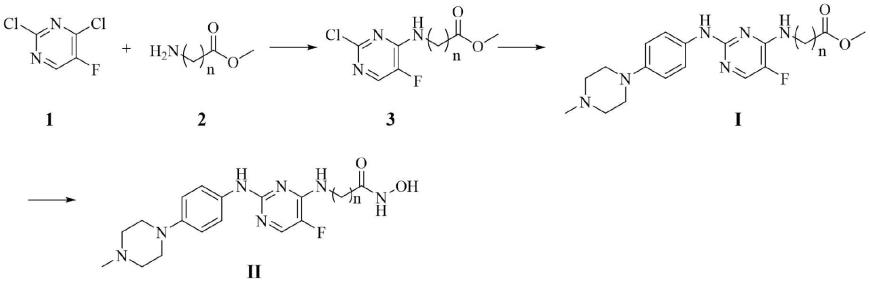
在本发明的第二方面,本发明还提供了上述哌嗪苯基氨基取代的嘧啶氨基酸衍生物的制备方法,所述制备方法包括进行如下反应路线:
[0023][0024]
其中,n为1,2,3,4,5,6,7;
[0025]
试剂及条件:(a)n,n-二异丙基乙胺(dipea),异丙醇,85℃,4h;(b)4-甲基哌嗪-1-基苯胺,三氟乙酸,正丁醇,110℃,12h;(c)氢氧化钾,盐酸羟胺,无水甲醇,0℃~r.t.,2h。
[0026]
具体步骤如下:
[0027]
(1)化合物1与氨基酸甲酯2缩合得中间体3。
[0028]
(2)中间体3在三氟乙酸条件下与4-甲基哌嗪-1-基苯胺缩合得哌嗪苯基氨基取代的嘧啶氨基酸衍生物i。
[0029]
(3)哌嗪苯基氨基取代的嘧啶氨基酸衍生物i与羟胺钾溶液反应得哌嗪苯基氨基取代的嘧啶氨基酸衍生物ii。
[0030]
(4)哌嗪苯基氨基取代的嘧啶氨基酸衍生物i的制备方法如下:
[0031]
(i)将化合物1和氨基酸甲酯2溶于异丙醇中,加入dipea,85℃反应4小时。tlc检测,反应完全,冷却至室温,有大量固体析出,过滤,滤饼用乙酸乙酯重结晶,得中间体3。
[0032]
(ii)将中间体3溶于正丁醇中,加入4-甲基哌嗪-1-基苯胺,再向溶液中滴加三氟乙酸,110℃反应12h。tlc检测,反应完全,冷却至室温,减压蒸除溶剂,硅胶柱层析,得哌嗪苯基氨基取代的嘧啶氨基酸衍生物i。
[0033]
(5)哌嗪苯基氨基取代的嘧啶氨基酸衍生物ii的制备方法如下:
[0034]
利用氢氧化钾和盐酸羟胺及无水甲醇制备nh2ok溶液。将哌嗪苯基氨基取代的嘧啶氨基酸衍生物i溶于nh2ok溶液中,室温反应2h。tlc检测,反应完全,减压蒸除溶剂,加水,用稀盐酸调ph值至6-7,有固体析出,过滤,滤饼用甲醇或乙酸乙酯重结晶,得哌嗪苯基氨基取代的嘧啶氨基酸衍生物ii。
[0035]
在本发明的第三方面,本发明还提供了一种药物组合物,其含有上述哌嗪苯基氨基取代的嘧啶氨基酸衍生物或其药学上可接受的盐。
[0036]
本发明化合物的药物组合物,可以选自以下任意方式施与:口服、喷雾吸入、直肠给药、鼻腔给药、阴道给药、局部给药、非肠道给药如皮下、静脉、肌内、腹膜内、鞘内、心室内、胸骨内或颅内注射或输入,或借助一种外植的储器用药,其中优选口服、肌注、腹膜内或静脉内用药方式。
[0037]
在本发明的第四方面,本发明还提供了一种药物制剂,其包含上述哌嗪苯基氨基取代的嘧啶氨基酸衍生物或其药学上可接受的盐或含有上述哌嗪苯基氨基取代的嘧啶氨基酸衍生物或其药学上可接受的盐的组合物和药学上可接受的辅料和/或载体。
[0038]
本发明哌嗪苯基氨基取代的嘧啶氨基酸衍生物或含有它的药物组合物可以单位剂量形式给药。给药剂型可以是液体剂型、固体剂型。液体剂型可以是真溶液类、胶体类、微粒剂型、乳剂剂型、混旋剂型。其他剂型例如片剂、胶囊、滴丸、气雾剂、丸剂、粉剂、溶液剂、混悬剂、乳剂、颗粒剂、栓剂、冻干粉针剂、包合物、填埋剂、贴剂、擦剂等。
[0039]
本发明的药物组合物或药物制剂中还可以含有常用的载体,这里所述可药用载体包括但不局限于:离子交换剂,氧化铝,硬脂酸铝,卵磷脂,血清蛋白如人血清蛋白,缓冲物质如磷酸盐,甘油,山梨酯,山梨酸钾,饱和植物脂肪酸的部分甘油酯混合物,水,盐或电解质,如硫酸鱼精蛋白,磷酸氢二钠,磷酸氢钾,氯化钠,锌盐,胶态氧化硅,三硅酸镁,聚乙烯吡咯烷酮,纤维素物质,聚乙二醇,羧甲基纤维素钠,聚丙烯酸酯,蜂蜡,羊毛酯等。载体在药物组合物中的含量可以是1重量%-98重量%,通常大约占到80重量%。为方便起见,局部麻醉剂,防腐剂,缓冲剂等可直接溶于载体中。
[0040]
口服片剂和胶囊可以含有赋形剂如粘合剂,如糖浆,阿拉伯胶,山梨醇,黄芪胶,或聚乙烯吡咯烷酮,填充剂,如乳糖,蔗糖,玉米淀粉,磷酸钙,山梨醇,氨基乙酸,润滑剂,如硬脂酸镁,滑石,聚乙二醇,硅土,崩解剂,如马铃薯淀粉,或可接受的增润剂,如月桂醇钠硫酸盐。片剂可以用制药学上公知的方法包衣。
[0041]
口服液可以制成水和油的悬浮液,溶液,乳浊液,糖浆,也可以制成干品,用前补充水或其它合适的媒质。这种液体制剂可以包含常规的添加剂,如悬浮剂,山梨醇,纤维素甲醚,葡萄糖糖浆,凝胶,羟乙基纤维素,羧甲基纤维素,硬脂酸铝凝胶,氢化的食用油脂,乳化剂,如卵磷脂,山梨聚糖单油酸盐,阿拉伯树胶;或非水载体(可能包含可食用油),如杏仁
hcl调ph值至6-7,有固体析出,过滤,滤饼用甲醇/乙酸乙酯重结晶,得哌嗪苯基氨基取代的嘧啶氨基酸衍生物ii。
[0066]
ii-1:3-((5-氟-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)氨基)-n-羟基丙酰胺
[0067]1h nmr(400mhz,dmso-d6)δ10.38(s,1h),8.72(s,2h),7.74(d,j=3.7hz,1h),7.49(d,j=9.1hz,2h),7.27(t,j=5.9hz,1h),6.76(d,j=9.1hz,2h),3.53-3.48(m,2h),3.02
–
2.91(m,4h),2.40
–
2.33(m,4h),2.26(t,j=7.1hz,2h),2.14(s,3h).
13
c nmr(101mhz,dmso-d6)δ167.85,156.52(d,j=2.4hz),152.37(d,j=12.2hz),145.93,141.13(d,j=245hz),139.06(d,j=18.4hz),134.17,119.77,116.41,55.24,49.54,46.28,37.03,32.48.hrms(esi)m/z calcdfor c
18h25
fn7o2[m+h]
+
390.2054,found 390.2040.
[0068]
ii-2:4-((5-氟-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)氨基)-n-羟基丁酰胺
[0069]1h nmr(400mhz,dmso-d6)δ10.35(s,1h),8.68(s,1h),8.67(s,1h),7.73(d,j=3.7hz,1h),7.47(d,j=9.1hz,2h),7.34(t,j=5.6hz,1h),6.77(d,j=9.1hz,2h),3.01
–
2.91(m,4h),2.39
–
2.33(m,4h),2.14(s,3h),1.97(t,j=7.5hz,2h),1.77-1.70(m,2h).
13
c nmr(101mhz,dmso-d6)δ169.27,156.54(d,j=2.4hz),152.50(d,j=12.2hz),145.92,141.10(d,j=241hz),138.96(d,j=18.5hz),134.21,119.72,116.40,55.24,49.53,46.28,30.44,25.44.hrms(esi)m/z calcd for c
19h27
fn7o2[m+h]
+
404.2210,found404.2196.
[0070]
ii-3:5-((5-氟-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)氨基)-n-羟基戊酰胺
[0071]1h nmr(400mhz,dmso-d6)δ10.36(s,1h),8.76(s,1h),8.67(s,1h),7.79(d,j=3.8hz,1h),7.55(d,j=9.1hz,2h),7.41(t,j=5.4hz,1h),6.84(d,j=9.1hz,2h),3.11-3.03(m,4h),2.65
–
2.57(m,4h),2.33(s,3h),2.04
–
1.93(m,2h),1.62
–
1.48(m,4h).
13
c nmr(101mhz,dmso-d6)δ169.48,156.54(d,j=2.3hz),152.49(d,j=12.3hz),145.58,141.11(d,j=241hz),138.88(d,j=17.9hz),134.43,119.77,116.50,54.72,49.01,45.49,32.61,29.07,23.28.hrms(esi)m/z calcd for c
20h29
fn7o2[m+h]
+
418.2367,found418.2356.
[0072]
ii-4:6-((5-氟-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)氨基)-n-羟基己酰胺
[0073]1h nmr(400mhz,dmso-d6)δ10.34(s,1h),8.71(d,j=32.0hz,2h),7.78(d,j=3.8hz,1h),7.55(d,j=9.0hz,2h),7.36(t,j=5.0hz,1h),6.83(d,j=9.0hz,2h),3.43
–
3.35(m,overlapped with water,2h),3.05(s,4h),2.57
–
2.51(m,overlapped with dmso,4h),2.27(s,3h),1.95(t,j=7.3hz,2h),1.66
–
1.46(m,4h),1.30(dd,j=15.1,8.3hz,2h).
13
c nmr(101mhz,dmso-d6)δ169.53,156.57(d,j=2.5hz),152.50(d,j=12.2hz),145.81,141.10(d,j=242hz),138.87(d,j=18.5hz),134.32,119.77,116.40,55.05,49.33,45.98,32.78,30.86,29.12,26.65,25.44.hrms(esi)m/z calcd for c
21h31
fn7o2[m+h]
+
432.2523,found432.2514.
[0074]
ii-5:7-((5-氟-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)嘧啶-4-基)氨基)-n-羟基
庚酰胺
[0075]1h nmr(400mhz,dmso-d6)δ10.33(s,1h),8.72(d,j=24.7hz,2h),7.78(d,j=3.8hz,1h),7.55(d,j=9.0hz,2h),7.37(t,j=5.5hz,1h),6.82(d,j=9.1hz,2h),3.33-3.27(m,2h),3.09
–
2.98(m,4h),2.49-2.47(m,4h),2.24(s,3h),1.94(t,j=7.3hz,2h),1.59-1.46(m,4h),1.37
–
1.23(m,4h).
13
c nmr(101mhz,dmso-d6)δ169.57,156.56(d,j=2.4hz),152.50(d,j=12.2hz),145.83,141.10(d,j=241hz),138.86(d,j=19.1hz),134.32,119.76(s,2c),116.37(s,2c),55.09,49.41,46.06,32.74,29.26,28.93,26.79,25.68.hrms(esi)m/z calcd for c
22h33
fn7o2[m+h]
+
446.2680,found446.2669.
[0076]
实验例:目标化合物对flt3和hdac抑制活性测试、对肿瘤细胞的抗增殖活性测试实验
[0077]
此处目标化合物为本发明实施例提供的实施例2和实施例3制备得到的哌嗪苯基氨基取代的嘧啶氨基酸衍生物i和哌嗪苯基氨基取代的嘧啶氨基酸衍生物ii。
[0078]
1)目标化合物对flt3抑制活性实验:
[0079]
实验材料和仪器:flt3激酶抑制活性实验由英国公司eurofins pharma协助完成。
[0080]
实验方法:将所有测试的本发明化合物使用dmso配制成最终测试浓度50倍的工作液。首先将化合物工作液作为第一组分加入到测试孔中,然后加入激酶buffer稀释的flt3激酶溶液。mg/atp的加入,激酶反应被引发。随后,室温下孵育40分钟,加入0.5%的磷酸溶液终止反应。取10μl反应液点到p30滤纸垫上,使用0.425%磷酸洗涤4次,每次洗涤4分钟,然后用甲醇洗涤一次,随后进行干燥和闪烁计数。
[0081]
试验中设定了化合物测试组(c),阳性对照组(p)和空白对照组(b)。阳性对照组中不加测试化合物,使用dmso代替(最终浓度2%),其他组分与测试组相同(残留激酶活性100%);空白对照组中使用星形孢菌素(staurosporine)代替测试化合物,消除激酶活性并建立基线(残留激酶活性0%)。
[0082][0083]
使用gragphad prism6.0软件,以浓度对数为横坐标,抑制率为中坐标,拟合曲线,计算ic
50
值。目标化合物对btk、flt3激酶抑制活性测定实验结果见表1。
[0084]
表1.目标化合物对flt3抑制活性
[0085][0086][0087]
ic
50
:半数抑制浓度
[0088]
a:ic
50
《1μm;b:1μm《ic
50
《10μm;c:10μm《ic
50
[0089]
表1实验数据表明,大部分化合物对flt3具有强效的抑制活性(ic
50
《1μm),其ic
50
值显著低于阳性对照药tandutinib。
[0090]
2)化合物对hdac抑制活性实验:
[0091]
实验材料和方法:boc-lys(acetyl)-amc(hela细胞核提取荧光底物)购于瑞士bachemag公司,tris-hcl、trypsin和edta购于sigma-aldrich公司,tsa购于阿拉丁科技生化有限公司,甘油、nacl、96孔平底荧光板购于鸿飞试剂公司。
[0092]
缓冲液:15mm tris-hcl(ph8.0),250mm nacl,250μm edta,10%甘油
[0093]
hdac酶溶液:按试验要求用buffer稀释。
[0094]
荧光底物溶液:底物用dmso溶解配成30mm储备液于-20℃保存,使用时用hdac buffer稀释至300μm。
[0095]
终止液:10mg/ml trypsin,50mm tris-hcl(ph8.0),100mm nacl,2μm tsa。
[0096]
酶标仪:varioskan flash光谱扫描多功能读数仪。
[0097]
实验步骤:
[0098]
100%对照组:将10μl hdacs酶溶液与50μl hdac buffer混合,在37℃下孵育5min,加入40μl底物溶液后,继续在37℃孵育30min,加入100μl终止液,继续在37℃孵育20min,使用酶标仪测定反应液在390nm/460nm的荧光强度,数值为100%组荧光度。
[0099]
空白对照组:将40μl底物溶液与60μl hdac buffer混合,在37℃下孵育30min,加入100μl终止液后,继续在37℃孵育20min,使用酶标仪测定反应液在390nm/460nm的荧光强度,数值为空白组的荧光度。
[0100]
实验组:将10μl hdacs酶溶液与50μl用hdac buffer稀释的待测化合物混合,在37℃下孵育5min,加入40μl底物溶液后,继续在37℃孵育30min,加入100μl终止液后,继续在37℃孵育20min,使用酶标仪测定反应液390nm/460nm的荧光强度,数值为待测化合物该浓度的荧光度。
[0101]
根据公式计算不同浓度下的抑制率,用origin软件进行s曲线拟合,计算待测本发明化合物ic
50
值,如表2所示。
[0102]
表2.目标化合物对hdac抑制活性
[0103][0104]
a:ic
50
《1μm;b:1μm《ic
50
《10μm;c:10μm《ic
50
[0105]
表2实验数据表明,化合物ii-3、ii-4和ii-5对hdac1和hdac6显示出强效的抑制作用(ic
50
《1μm),其ic
50
值与阳性药saha的ic
50
值相当。值得注意的是,化合物ii-3、ii-4和ii-5对hdac和flt3表现出双重抑制作用。
[0106]
3)化合物对肿瘤细胞的生长抑制活性实验:
[0107]
实验材料和仪器:血液系统恶性肿瘤细胞jeko-1、mv4-11、k562、z138、hel和molt4细胞株、rpmi-1640培养基、胎牛血清、pbs缓冲液、青霉素纳(10000units/ml)-硫酸链霉素(10mg/ml)、cck-8试剂盒、倒置光学显微镜、细胞培养箱、超净工作台、台式离心机、酶标仪、超低温冰箱。
[0108]
实验方法:
[0109]
取对数生长期肿瘤细胞接种于96孔培养板中,细胞数为1
×
104/孔,加入不同浓度所测本发明化合物的细胞培养液,同时设立阳性对照组和dmso空白对照组,调整dmso浓度≤1
‰
。每个浓度设3个复孔,加毕,置37℃,5%co2恒温培养箱中孵育72h。随后每孔加入20μl cck-8溶液,培养板置于37℃,5%co2恒温培养箱中继续孵育1-4h,用酶标仪测定其在450nm波长下的吸光度值,所得数值与阴性dmso对照组进行归一化处理,应用prism 6.0软件计算ic
50
值
[0110][0111]
表3.代表性化合物对血液系统恶性肿瘤细胞的抗增殖作用
[0112][0113]
a:ic
50
《2μm;b:2μm《ic
50
《20μm;c:20μm《ic
50
[0114]
我们选择具有强效hdac/flt3双重抑制剂活性的代表性化合物,进一步研究了其对血液系统恶性肿瘤细胞的抗增殖活性,结果如表3所示,化合物ii-3、ii-4和ii-5对jeko-1、mv4-11和molt4细胞显示出强效的抗增殖活性,特别是化合物ii-5对所测的6株细胞的抗增殖活性与阳性药saha相当甚至更优。
[0115]
以上所述仅为本公开的优选实施例而已,并不用于限制本公开,对于本领域的技术人员来说,本公开可以有各种更改和变化。凡在本公开的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本公开的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1