一种二氯喹啉酸的制备方法与流程
1.本发明涉及农药合成技术领域,特别是涉及一种二氯喹啉酸的制备方法。
背景技术:
2.二氯喹啉酸又名3,7-二氯-8-喹啉羧酸,是一种药效显著的水稻田用除草剂,该药用于直播稻田和移栽稻田防治稗草属、田角皂属、臂形草属、鸭舌草等,尤其对稗草效果显著,对作物安全,施药时间不受杂草生长阶段限制,属激素型喹啉酸类除草剂,其结构式如下:
[0003][0004]
该除草剂具有除草效果好,对作物安全,施药时间不受杂草生长阶段限制等优点,被市场广泛认可。
[0005]
美国专利us4632696以及us4497651指出二氯喹啉酸的合成过程中二氯喹啉酸中羧基的获得,是通过二氯化合物与强酸,如浓盐酸和浓硫酸在50~150℃反应,但并未详细指明氧化合成工艺及其氧化剂。
[0006]
专利申请cn101851197a公开了一种二氯喹啉羧酸的合成和精制方法,以苯胺和甘油为主要原料,经缩合、氯化、氧化水解等三步合成的工艺路线。具体步骤为:在硫酸介质中,经催化剂催化作用,苯胺与甘油缩合反应,得到喹啉;喹啉在二氯苯溶液中,经引发剂作用,通氯氯化,得到7-氯-8-甲基喹啉的氯化物的二氯苯溶液,脱除二氯苯后,加水,加硫酸,得到氧化配制液,经硝酸氧化,得到3,7-二氯-8-喹啉羧酸。其反应方程式如下:
[0007][0008]
二氯喹啉酸氯化工艺中,除了生成以上方程式中所示的氯化中间产物外,通常会伴随有如下所示的少量氯化杂质生成,这些杂质会带到最终产品中,从而影响产品含量,甚至在市售产品中可检测到环上3-4个氯的杂质。
[0009][0010]
现有二氯喹啉酸氧化工艺,采用在浓硫酸介质中,滴加硝酸溶液氧化7-氯-8-甲基喹啉的氯化物,滴加与保温所需时间在20~24小时,是缩合工序或氯化工序所需时间的2.5~3倍,故产能的扩大受到氧化工序的制约,同时,使用硝酸作为氧化剂时,投入量很大,一吨二氯喹啉酸产品需要消耗98%浓硝酸1.5吨,从而产生大量的硝硫混酸废酸,不易处理,混合废酸处理已成为该产品发展壮大的瓶颈,另外,使用硝酸作为氧化剂,产品中会产生硝基物,影响产品质量。目前市售产品含量大多在87%~92%之间,需经过除硝基、重结晶等复杂的提纯程序方可将产品含量提升至97%以上,但同时大大增加了成本。该方法虽然每吨产品产生大约40-50吨的废水,但仍然为目前市场上所采用的主流的生产方法。
[0011]
cn111377862公开了一种二氯喹啉酸的制备方法,该方法包括以下步骤,1)以n-羟基邻苯二甲酰亚胺类化合物和偶氮二异丁腈为催化剂,以氧气为氧化剂,将7-氯-8-甲基喹啉氧化得到7-氯-8-喹啉羧酸;2)以偶氮二异丁腈为催化剂,使7-氯-8-喹啉羧酸和氯气进行氯化反应得到3,7-二氯-8-喹啉羧酸。反应方程式如下:
[0012][0013]
此方法由于有羧基存在,钝化了苯环,氯化单一的上到了n杂环上,而且通入氯气不用大大过量,所以产品的含量得以大大提升,但前面氧化反应在乙腈为溶剂中,纯氧气的压力要4-10mpa,反应温度在80-100℃,由于压力过高,温度又高,乙腈闪点低并易燃易爆,从而大大增加了反应的安全风险,不适于大规模商业化生产。
[0014]
chem.commun.2002,180
–
181也报道这种喹啉环上的甲基氧化,在低的压力下不反应。
[0015]
cn103420909报道了另外一种氧化方法,采用昂贵的co-mn-br三元mc催化体系,以脂肪酸为溶剂,在反应温度为155~205℃,反应压力为1.0~1.8mpa的纯氧条件下氧化得到目标产物二氯喹啉酸,方程式如下:
[0016][0017]
该方法虽然降低了反应压力,但是氯化杂质问题并没有解决,并且该方法要求氧化的起始原料中一氯产物的比率要大于90%,否则反应很难发生,而在实际生产氯化过程中,氯气是过量的,并且在一氯产物大于50%时再继续氯化就会伴随有二氯甚至三氯及多氯产物生成,很难控制,故也不适于商业化生产。
[0018]
在其它可查询到的专利中,都是在氯化后,变换氧化方法,有用次氯酸钠、高氯酸、双氧水等替代硝酸氧化,但由于这些氧化剂的氧化性较弱,所得产品收率偏低,虽然解决了硝硫混酸问题,但废水量并没有本质上的减少,也没有解决氯化杂质问题,故也很少见应用于商业化生产。
[0019]
综上所述,研发一种操作简便、高收率、高质量,并从本质上能够减少合成工艺中大量废酸和废水的产生,且低安全风险的合成工艺,具有非常重要的意义。
技术实现要素:
[0020]
为了解决上述工业化中的问题,本发明提供一种操作简便、低安全风险、高收率、高含量的二氯喹啉酸的制备方法。
[0021]
具体而言,本技术提供了如下技术方案:
[0022]
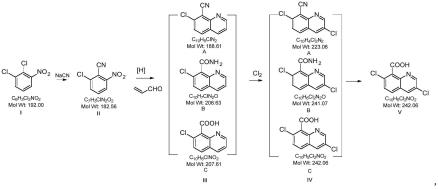
一种二氯喹啉酸的制备方法,采用如下路线:
[0023][0024]
包括以下步骤:
[0025]
(1)在溶剂中,在反应温度为50~300℃下,将式ⅰ所示化合物在催化剂存在下与氰化钠进行反应,生成式ⅱ所示化合物;
[0026]
(2)在反应温度为-20~200℃下,将式ⅱ所示化合物和丙烯醛在有机溶剂和盐酸或硫酸中在催化剂下,或加氢还原并环合同步进行一锅法合成式ⅲ所示三种结构的混合化合物;
[0027]
(3)在反应温度为10~200℃下,向式ⅲ所示结构化合物存在的有机溶剂中,在催化剂存在下通入氯气,生成式ⅳ所示结构化合物;
[0028]
(4)在反应温度为10~200℃下,式ⅳ所示结构化合物在酸性或碱性条件下水解,生成式
ⅴ
所示的二氯喹啉酸。
[0029]
其中,所述步骤(2)中,其还原中间产物包括以下三种,结构式分别为:其中,所述步骤(2)中,其还原中间产物包括以下三种,结构式分别为:反应过程如下:
[0030][0031]
其中,式ⅵ、式ⅶ、式
ⅷ
所示结构的化合物均可与丙烯醛发生关环反应,故此处采用一锅法进行反应;
[0032]
或者,步骤(2)也可以通过高压釜加氢得到单一的式
ⅷ
所示结构化合物,然后再进行关环反应,通过如下反应式方式进行:
[0033][0034]
其中,所述步骤(1)中所述溶剂为2,3-二氯硝基苯、苯甲醚、苯乙醚、丁基苯基醚、
二甲苯、三甲苯、二甘醇二甲醚、二甘醇二乙醚、邻硝基苯甲醚、n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dmac)、n,n-二乙基甲酰胺(defa)、二甲基亚砜(dmso)、二甲基砜(dmso2)、环丁砜、2,4-二甲基环丁砜、n-甲基吡咯烷酮(nmp)、六甲基磷酸三酰胺(hmpa)、1,3-二甲基-2-咪唑啉酮(dmi)中的一种或多种;
[0035]
优选为2,3-二氯硝基苯、n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dmac)、n,n-二乙基甲酰胺(defa)、二甲基亚砜(dmso)、聚乙二醇、二甲基砜(dmso2)、环丁砜、2,4-二甲基环丁砜、n-甲基吡咯烷酮(nmp)、六甲基磷酸三酰胺(hmpa)、1,3-二甲基-2-咪唑啉酮(dmi)中的一种或多种;
[0036]
更优选为2,3-二氯硝基苯、n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dmac)、n,n-二乙基甲酰胺(defa)、二甲基亚砜(dmso)、环丁砜、n-甲基吡咯烷酮(nmp)、六甲基磷酸三酰胺(hmpa)、1,3-二甲基-2-咪唑啉酮(dmi)中的一种或多种。
[0037]
其中,步骤(1)中溶剂的用量优选为式ⅰ所示化合物重量1~10倍,更优选为1~5倍;
[0038]
其中,所述步骤(1)中所述催化剂为溴化钠、溴化钾、碘化钠、碘化钾、溴化镍、三乙烯二胺、冠醚、氰化亚铜、四丁基氯化铵、四丁基溴化铵、苄基三乙基氯化铵、苄基三乙基溴化铵、氯化亚铜、n,n-二甲基苄胺、4-二甲氨基吡啶、二氮杂二环(dbu)、n,n-二甲基苯胺、四丁基溴化磷、苄基三苯基氯化磷中的一种或多种;
[0039]
优选为碘化钾、溴化镍、三乙烯二胺、冠醚、氰化亚铜、四丁基溴化铵、苄基三乙基溴化铵、氯化亚铜、n,n-二甲基苄胺、4-二甲氨基吡啶、二氮杂二环(dbu)、n,n-二甲基苯胺、四丁基溴化磷、苄基三苯基氯化磷中的一种或多种;
[0040]
更优选为氰化亚铜、四丁基溴化铵、四丁基溴化磷、苄基三苯基氯化磷、三乙烯二胺、冠醚中的一种或多种。
[0041]
其中,所述步骤(1)中所述催化剂的用量为式ⅰ所示化合物摩尔量的0.001~1倍;优选为0.01~0.5倍;更优选为0.02~0.1倍;所述步骤(1)中的反应温度为50~300℃,优选为100~250℃,更优选为150~200℃。
[0042]
步骤(1)中所述氰化钠的用量优选为式ⅰ所示化合物摩尔量的0.5~2倍;更优选为0.9~1.5倍。
[0043]
其中,所述步骤(2)中所述溶剂为2,3-二氯硝基苯、硝基苯、苯甲醚、苯乙醚、丁基苯基醚、二甲苯、三甲苯、二甘醇二甲醚、二甘醇二乙醚、邻硝基苯甲醚,二氯甲烷、二氯乙烷、氯仿、四氯化碳、甲苯、氯苯、二氯苯、邻氯甲苯、乙二醇二甲醚、乙酸甲酯、乙酸乙酯、乙酸丁酯、硝基甲烷、硝基乙烷、硝基苯、硝基甲苯、乙腈、四氢呋喃、甲基四氢呋喃、甲基叔丁基醚、乙二醇二乙醚、三氟乙酸中的一种或多种;
[0044]
优选为2,3-二氯硝基苯,硝基苯、二氯甲烷、二氯乙烷、甲苯、二甲苯、氯苯、二氯苯、邻氯甲苯、乙酸乙酯、乙酸丁酯、三氟乙酸的一种或多种;
[0045]
更优选为二氯甲烷、二氯乙烷、硝基苯、甲苯、二甲苯、氯苯、二氯苯、三氟乙酸的一种或多种。
[0046]
其中,步骤(2)中溶剂的用量优选为式ⅱ所示化合物重量的1~10倍,更优选为1~5倍;
[0047]
所述步骤(2)中所述催化剂为铁、镁、铝、锌、锡中的一种或多种;
[0048]
所述加氢还原采用的催化剂为钯碳、铂碳、雷尼镍、钌中的一种或多种。
[0049]
所述步骤(2)中的反应温度为0~300℃,优选为30~150℃,更优选为30~100℃。
[0050]
所述步骤(2)中所述催化剂的用量为式ⅱ所示化合物摩尔量的0.01~2倍;优选为0.1~1.5倍;更优选为0.5~1.2倍。
[0051]
所述步骤(2)中所述丙烯醛的用量优选为式ⅱ所示化合物摩尔量的0.5~2倍;更优选为0.8~1.5倍。
[0052]
其中,所述步骤(3)中所述溶剂优选为二氯苯、c
1-c5的饱和脂肪酸等;更优选为二氯苯、乙酸等;溶剂的用量优选为式ⅲ所示化合物重量的1~10倍,更优选为1~5倍。
[0053]
所述步骤(3)中所述催化剂为过氧化环己酮、过氧化二苯甲酰、过氧化苯甲酰、叔丁基过氧化氢、偶氮二异丁腈、偶氮二异庚腈、四丁基氯化铵、四丁基溴化铵、苄基三乙基氯化铵、苄基三乙基溴化铵中的一种或多种;
[0054]
优选为过氧化苯甲酰、偶氮二异丁腈、四丁基溴化铵、苄基三乙基氯化铵、苄基三乙基溴化铵中的一种或多种;
[0055]
更优选为偶氮二异丁腈和/或四丁基溴化铵。
[0056]
其中,所述步骤(3)中所述催化剂的用量为式ⅲ所示化合物摩尔量的0.001~2倍;优选为0.01~1.0倍;更优选为0.05~0.5倍。
[0057]
步骤(3)中所述氯气的用量优选为式ⅲ所示化合物摩尔量的0.5~2倍;更优选为0.8~1.3倍。
[0058]
所述步骤(3)中的反应温度为0~300℃,优选为50~200℃,更优选为80~160℃。
[0059]
其中,所述步骤(4)中的酸为磷酸、多聚磷酸、硝酸、硫酸、盐酸、甲基磺酸、对甲苯磺酸、草酸中的一种或多种;优选为磷酸、硝酸、硫酸、盐酸中的一种或多种,更优选为硝酸、硫酸、盐酸中的一种或多种。所述酸的用量优选为式ⅳ所示结构化合物摩尔量的0.5~10倍。
[0060]
其中,所述步骤(4)中的碱为氢氧化锂、氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、氨水或c
1-c4的醇钠或醇碱中的一种或多种;优选为氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、甲醇钠、乙醇钠、叔丁醇钠、叔丁醇钾、甲醇钾、乙醇钾中的一种或多种;更优选为氢氧化钠、氢氧化钾、甲醇钠、乙醇钠、叔丁醇钠中的一种或多种。所述碱的用量优选为式ⅳ所示结构化合物摩尔量的0.5~10倍,更优选为1~5倍。
[0061]
与现有技术相比,本发明的二氯喹啉酸的制备方法至少具有以下有益效果:
[0062]
(1)本发明首次发现并公开了采用非氧化方式制备二氯喹啉酸的工艺,确定了适宜的工艺条件。
[0063]
(2)本发明的反应体系内杂质少,反应及后处理工艺大为简化,粗品经简单提纯即可得到含量97%以上的产品。
[0064]
(3)本发明工艺避免了传统工艺中采用硝酸氧化,从而产生大量的硝硫混酸废酸,从而从本质上降低了废水的排放量和处理难度。
[0065]
(4)本发明工艺避免了采用高温高压氧气的氧化方式,从本质避免的价格昂贵的氧化催化剂的使用,在降低成本的同时,也从本质上降低了反应的安全风险。
[0066]
下面结合附图对本发明的二氯喹啉酸的制备方法作进一步说明。
附图说明
[0067]
图1为实施例2-1的产物的液相图谱;
[0068]
图2为最终产物二氯喹啉酸的液相图谱。
具体实施方式
[0069]
下面结合具体实施例来详述本发明,以便于更好地理解本发明的实质内涵,但本发明绝非仅限于这些实施例。其中所用化学原料均为市售工业品。
[0070]
一种二氯喹啉酸的制备方法,采用如下路线:
[0071][0072]
实施例1
[0073]
上述合成路线中,式ⅱ所示结构化合物的合成:
[0074]
室温下,将2,3-二氯硝基苯、氰化钠和氰化亚铜投入到1000ml反应瓶中,缓慢用油浴加热升温至90℃,待2,3-二氯硝基苯熔化后得到浆状物,开启搅拌,继续升温,升温至100℃后,加入dmf,继续升温至160℃,保温反应6小时,然后再继续升温至170℃,保温反应8小时,取样分析,检测到2,3-二氯硝基苯小于0.5%,反应完毕,降温至90℃,负压脱溶回收dmf,脱溶结束后向反应瓶中加入氯苯,搅拌充分溶解后在80℃过滤除去无机盐,滤液在70℃用5%的氨水搅拌洗涤1h,静置、分层,有机相再用水洗一次。再次分层后有机相直接去还原反应。
[0075]
下表中为投料配比对反应收率的影响:
[0076][0077][0078]
实施例2
[0079]
前述合成路线中,式ⅲ所示结构化合物的合成:
[0080]
实施例2-1
[0081]
向1l反应瓶中加入37g(0.2mol)2-硝基-6-氯苯腈、100ml甲醇、8g铁粉,开启搅拌升温,控制温度在50-55℃,缓慢滴加60ml30%h2so4,滴加结束后升温至回流,在回流状态下保温反应1h,取样检测cnbn《0.5%停止反应。然后向反应体系中加入硝基苯100g,然后升温至70℃,滴加丙烯醛11.5g(0.2mol),3小时滴完。滴完后保温搅拌反应1小时后,升温至125度,保温反应6小时,同时回收甲醇,反应结束后,降温至50度,将反应液用10%的naoh水溶液调节ph至3.5,抽滤,用水淋洗滤饼,得式ⅲ所示结构化合物的混合物湿品(以铁粉还原得到产物以氰基形式为主),液相图谱如图1所示,滤液分层后硝基苯回收套用。
[0082]
实施例2-2
[0083]
向1l高压釜中加入37g(0.2mol)2-硝基-6-氯苯腈、100ml甲醇、0.5g钯碳,高压釜盖好盖后,用氮气置换3次,在用氢气置换三次,然后充氢气至0.8mpa,开启搅拌升温至80℃,继续充氢气至2.0mpa,开始吸氢,控制温度在80-100℃,压力在2.0-3.0mpa反应直至结束。降温,过滤除去催化剂,滤液负压脱溶回收甲醇,脱溶结束后瓶内剩余物即为式8化合物。向脱溶后剩余物的反应瓶中加入100ml甲苯,0.1mmol三氟乙酸,升温至80℃,缓慢滴加丙烯醛,滴加结束后在90-100℃保温反应,tlc监测反应终点,反应结束后用nahco3中和,过滤,滤饼以乙醇/dmf重结晶得式iii所示结构化合物(加氢还原所得式iii化合物以羧基形式为主),可直接进入下一步反应。
[0084]
实施例3
[0085]
前述合成路线中,式ⅳ所示结构化合物的合成:
[0086]
室温下,将实施例2-1所得固体湿品加入到1l反应瓶中,然后加入150ml二氯苯,开启搅拌升温至120-140℃,通过回流分水器脱出体系中的水分,待水分脱除结束后,加入aibn0.2g,在140℃缓慢通入氯气14g,通气结束后保温反应1h,结束后降温,反应瓶内残留物直接进入下一步反应。
[0087]
实施例4
[0088]
前述合成路线中,式
ⅴ
所示结构化合物的合成:
[0089]
向实施例3的反应液中加入40ml浓硫酸,将烧瓶放置于冰浴中冷却,将事先己量取好的10ml浓硝酸加入恒压滴液漏斗中并小心缓慢的滴入烧瓶中,在此过程中要保持内部温度不能够超过10℃。滴加完毕后,将四口烧瓶转移到油浴锅中,缓慢升高温度至100℃反应2小时。取样分析,反应结束后,冷却反应液,将其小心缓慢地倒入适量的冰水混合物,在此过程中要不断地剧烈搅拌,有固体析出,将混合物抽滤得白色固体,以甲醇打浆,抽滤、干燥得二氯喹啉酸,其液相图谱如图2所示。四步反应总收率可达65%-70%,产品纯度大于98%。
[0090]
以上所述的实施例仅仅是对本发明的优选实施方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案作出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1