DL-高半胱氨酸硫内酯盐酸盐的制备方法与流程
dl-高半胱氨酸硫内酯盐酸盐的制备方法
技术领域
1.本技术涉及生物医药技术领域,尤其涉及dl-高半胱氨酸硫内酯盐酸盐的制备方法。
背景技术:
2.西替沃酮是用于脑血管障碍后遗症及老年痴呆症等的重要医药原料,产品形态为白色结晶粉末。一般用dl-高半胱氨酸硫内酯盐酸盐与乙酰氯合成得到西替沃酮。
3.dl-高半胱氨酸硫内酯盐酸盐是一种重要的生化试剂和药物中间体,产品形态为白色晶状粉末。一般通过化学还原法合成,即通过金属锌或锡、盐酸与dl-高胱氨酸进行还原反应制得,使用该方法合成会产生大量的氢气、锌盐(锡盐)和大量的废水,造成严重环境污染,且操作过程有一定的危险性。
4.申请号为200710053041.7的中国专利文件公开了一种dl-蛋氨酸在硫酸溶液中脱甲基氧化偶联得到dl-高半胱氨酸硫内酯盐酸盐的方法,该合成方法产生大量的二甲硫醚,二氧化硫等有毒气体,并产生大量的高盐废水,容易造成环境污染,给生产企业带来较高的环保成本压力。
5.申请号为201710695613.5的中国专利文件公开了一种dl-蛋氨酸与金属钠发生还原反应得到dl-高半胱氨酸硫内酯盐酸盐的方法,该合成方法在反应过程中会产生氢气,危险性较大,操作过程非常繁琐,反应成本高。
技术实现要素:
6.本发明的目的是提供dl-高半胱氨酸硫内酯盐酸盐的合成方法,该方法以巯基丁酸为原料合成dl-高半胱氨酸硫内酯盐酸盐,经酸催化缩合、溴取代、氨基取代一系列反应,提供了一种工艺简便,易于纯化,质量可控,收率高的dl-高半胱氨酸硫内酯盐酸盐的合成新途径。
7.一方面,本技术提供了一种dl-高半胱氨酸硫内酯盐酸盐的制备方法,所述方法包括如下步骤:步骤一、将硫代丁内酯与卤取代试剂进行卤代反应,获得2-卤代硫代丁内酯;步骤二、将所述2-卤代硫代丁内酯与氨基取代试剂进行氨基取代反应,获得dl-高半胱氨酸硫内酯盐酸盐。
8.进一步的,所述卤取代试剂为溴素;所述硫代丁内酯和卤取代试剂的摩尔比为1:(1.2~2.0)。
9.优选的,所述硫代丁内酯和卤取代试剂的摩尔比为1:1.5。
10.在一种优选的实施方式中,上述硫代丁内酯与溴素的反应,溶剂的用量为每4 ml氯仿中溶解1 g硫代丁内酯。
11.进一步的,所述卤代反应中采用三溴化磷作为催化剂。
12.在一种优选的实施方式中,上述硫代丁内酯和三溴化磷的质量比为1:0.1。
13.进一步的,所述卤代反应条件为:以氯仿作为溶剂,0℃~20℃温度下,反应1~10 h。
14.优选的,所述步骤一中还包括萃取分液、减压浓缩、打浆、过滤干燥的步骤。
15.优选的,所述卤代反应条件为:0℃~10℃温度下,反应5 h。
16.在一种优选的实施方式中,上述步骤一中打浆为石油醚打浆,石油醚打浆时,石油醚的用量为每1 g硫代丁内酯使用5 ml石油醚。
17.进一步的,所述氨基取代试剂为氨基钠;所述2-卤代硫代丁内酯和氨基取代试剂的摩尔比为1:(1.2~1.5)。
18.优选的,所述2-卤代硫代丁内酯和氨基取代试剂的摩尔比为1:1.3。
19.进一步的,所述氨基取代反应条件为:以四氢呋喃为溶剂,20℃~50℃温度下,反应1~5 h。
20.优选的,所述氨基取代反应条件为:30℃~40℃温度下,反应2 h。
21.在一种优选的实施方式中,上述四氢呋喃溶剂的用量为,每5 ml四氢呋喃中溶解1 g 2-溴硫代丁内酯。
22.进一步的,所述步骤二中还包括调节ph的步骤,所述ph调节至1~7;所述ph调节试剂为酸性试剂,所述酸性试剂选自浓盐酸和浓硫酸中的一种或两种。
23.优选的,所述步骤二中还包括降温析晶、过滤干燥的步骤。
24.优选的,所述ph调节至5~6,所述ph调节试剂为浓盐酸。
25.进一步的,所述硫代丁内酯采用巯基丁酸进行缩合反应制备获得;所述缩合反应采用算作为催化剂,所述酸选自浓盐酸和浓硫酸中的一种或两种。
26.优选的,所述酸催化剂为浓盐酸。
27.进一步的,所述缩合反应条件为:以四氢呋喃为溶剂,10℃~30℃温度下,反应2~4 h;所述硫代丁内酯的制备方法还包括减压浓缩、打浆、过滤干燥的步骤。
28.进一步的,所述缩合反应条件为:20℃~25℃温度下,反应3 h。
29.在一种优选的实施方式中,上述巯基丁酸在催化剂下的反应,溶剂和催化剂的用量为每5 ml四氢呋喃中加入2 ml浓盐酸,溶解1 g巯基丁酸。
30.并且,上述缩合反应中还包括减压浓缩、石油醚打浆的步骤,其中石油醚打浆时,石油醚的用量为每1 g巯基丁酸使用6 ml石油醚。
31.在一种优选的实施方式中,一种dl-高半胱氨酸硫内酯盐酸盐的制备方法,所述方法包括如下步骤:步骤一、取巯基丁酸、浓盐酸溶解在四氢呋喃中,控温20℃~25℃反应3 h,减压浓缩,石油醚打浆,过滤干燥得到硫代丁内酯。
32.步骤二、取硫代丁内酯、三溴化磷溶解在氯仿中,滴加溴素,控温0℃~10℃反应5 h,滴加水,搅拌分液,氯仿减压浓缩,石油醚打浆,过滤干燥得到2-溴硫代丁内酯。
33.步骤三、取2-溴硫代丁内酯溶解在四氢呋喃中,加入氨基钠控温30℃~40℃反应2 h,降温至10℃,滴加浓盐酸调节ph至5~6,控温低于20℃,降温至10℃析晶2 h,过滤干燥得到dl-高半胱氨酸硫内酯盐酸盐。
34.在一种优选的实施方式中,本技术还提供了一种西替沃酮的制备方法,所述方法
包括如下步骤:将所述的方法制备的dl-高半胱氨酸硫内酯盐酸盐中加入三乙胺作为催化剂,降温至-10℃~0℃,滴加乙酰氯进行反应,获得西替沃酮;所述dl-高半胱氨酸硫内酯盐酸盐和乙酰氯的摩尔比为1:(1.3~1.8);所述dl-高半胱氨酸硫内酯盐酸盐和乙酰氯的反应条件为:以二氯甲烷为溶剂,20℃~30℃温度下反应5~7 h;所述西替沃酮的制备方法还包括重结晶的步骤,重结晶后,乙酸乙酯溶解西替沃酮,活性炭脱色,滴加石油醚析晶,过滤干燥。
35.优选的,所述dl-高半胱氨酸硫内酯盐酸盐和乙酰氯的摩尔比为1:1.5。
36.在一种优选的实施方式中,上述dl-高半胱氨酸硫内酯盐酸盐和三乙胺的摩尔比为1:4,二氯甲烷、乙酸乙酯、石油醚的用量为每1 g dl-高半胱氨酸硫内酯盐酸盐加入10 ml二氯甲烷、2 ml乙酸乙酯或6 ml石油醚。
37.在一种优选的实施方式中,一种西替沃酮的制备方法,所述方法包括如下步骤:dl-高半胱氨酸硫内酯盐酸盐、三乙胺溶解在二氯甲烷中,降温至-10℃~0℃,滴加乙酰氯,滴加控温不超过0℃,滴加完毕后缓慢升温至常温反应6 h,饱和盐水洗涤有机相3次,萃取分层,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,剩余物乙酸乙酯溶解,活性炭脱色过滤,滴加石油醚析晶,0℃~10℃析晶4 h,过滤干燥得到西替沃酮。
38.本发明具有如下有益效果:1、本技术首次使用巯基丁酸为原料,制备dl-高半胱氨酸硫内酯盐酸盐,为dl-高半胱氨酸硫内酯盐酸盐的制备提供了新方法;2、本技术还提供了一种西替沃酮的制备方法,为西替沃酮的合成提供了一种新途径;3、本技术合成方法无需使用危险试剂,也无需加入金属钠、锌或锡等做还原剂,不会产生大量氢气、含锌或锡的废水,具有成本低、环境压力小、操作简单、危险性小、环保安全等优点,适合大规模生产。
附图说明
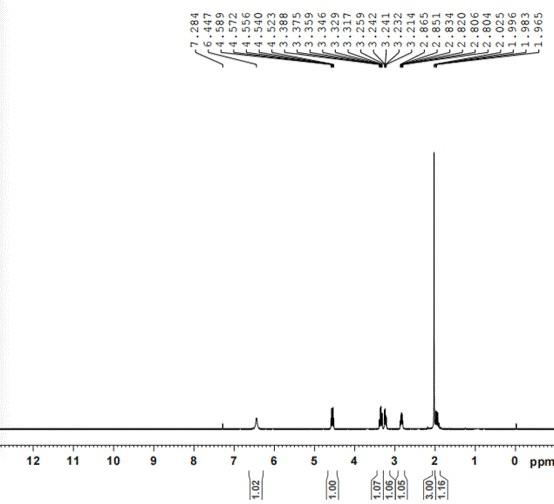
39.此处所说明的附图用来提供对本技术的进一步理解,构成本技术的一部分,本技术的示意性实施例及其说明用于解释本技术,并不构成对本技术的不当限定。在附图中:图1是西替沃酮的氢谱谱图;图2是西替沃酮的合成路径。
具体实施方式
40.为了更清楚的阐释本技术的整体构思,下面结合说明书附图以实施例的方式进行详细说明。在下文的描述中,给出了大量具体的细节以便提供对本发明更为彻底的理解。然而,对于本领域技术人员来说显而易见的是,本发明可以无需一个或多个这些细节而得以实施。在其他的例子中,为了避免与本发明发生混淆,对于本领域公知的一些技术特征未进行描述。
41.实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。
42.如未特殊说明,在以下实施方式中,所用试剂或仪器未注明生产厂商者,均为可以
通过市售购买获得的常规产品。
43.实施例1步骤一、取100 g(0.83 mol)巯基丁酸,200 ml浓盐酸溶解在500 ml四氢呋喃中,控温20℃~25℃反应3 h,减压浓缩四氢呋喃。600 ml石油醚打浆,过滤干燥得到75 g(0.73 mol)硫代丁内酯。
44.步骤二、取75 g(0.73 mol)硫代丁内酯,7.5 g三溴化磷溶解在300 ml氯仿中,滴加176 g(1.1 mol)溴素,控温0℃~10℃反应5 h,滴加水500 ml,搅拌分液,氯仿减压浓缩,375 ml石油醚打浆,过滤干燥得到120 g(0.66 mol)2-溴硫代丁内酯。
45.步骤三、取120 g(0.66 mol)2-溴硫代丁内酯溶解在600 ml四氢呋喃中,加入氨基钠33.6 g(0.86 mol)控温30℃~40℃反应2 h,降温至10℃,滴加浓盐酸调节ph至5~6,控温低于20℃,降温至10℃析晶2 h,过滤干燥得到91 g(0.59 mol)dl-高半胱氨酸硫内酯盐酸盐。
46.得到91 g dl-高半胱氨酸硫内酯盐酸盐,产品收率为71.08%,纯度:≥98.0%。
47.实施例2步骤一、取0.83 mol巯基丁酸,200 ml浓盐酸溶解在500 ml四氢呋喃中,控温20℃~25℃反应3 h,减压浓缩四氢呋喃。600 ml石油醚打浆,过滤干燥得到0.73 mol硫代丁内酯。
48.步骤二、取0.73 mol硫代丁内酯,7.5 g三溴化磷溶解在300 ml氯仿中,滴加0.949 mol溴素,控温0℃~10℃反应6 h,滴加水500 ml,搅拌分液,氯仿减压浓缩,375 ml石油醚打浆,过滤干燥得到0.64 mol2-溴硫代丁内酯。
49.步骤三、取0.64 mol 2-溴硫代丁内酯溶解在600 ml四氢呋喃中,加入氨基钠0.792 mol控温30℃~40℃反应3 h,降温至10℃,滴加浓盐酸调节ph至5~6,控温低于20℃,降温至10℃析晶2 h,过滤干燥得到dl-高半胱氨酸硫内酯盐酸盐。
50.得到90.2g dl-高半胱氨酸硫内酯盐酸盐,产品收率为70.48%,纯度:≥98.0%。
51.实施例3步骤一、取0.83 mol巯基丁酸,200 ml浓盐酸溶解在500 ml四氢呋喃中,控温20℃~25℃反应3 h,减压浓缩四氢呋喃。600 ml石油醚打浆,过滤干燥得到0.73 mol硫代丁内酯。
52.步骤二、取0.73 mol硫代丁内酯,7.5 g三溴化磷溶解在300 ml氯仿中,滴加1.241 mol溴素,控温0℃~10℃反应7 h,滴加水500 ml,搅拌分液,氯仿减压浓缩,375 ml石油醚打浆,过滤干燥得到0.64 mol2-溴硫代丁内酯。
53.步骤三、取0.64 mol 2-溴硫代丁内酯溶解在600 ml四氢呋喃中,加入氨基钠0.924 mol控温30℃~40℃反应4 h,降温至10℃,滴加浓盐酸调节ph至5~6,控温低于20℃,降温至10℃析晶2 h,过滤干燥得到dl-高半胱氨酸硫内酯盐酸盐。
54.得到86.45g dl-高半胱氨酸硫内酯盐酸盐,产品收率为67.53%,纯度:≥98.077.80%。
55.实施例4步骤一、取0.83 mol巯基丁酸,200 ml浓盐酸溶解在500 ml四氢呋喃中,控温20℃~25℃反应3 h,减压浓缩四氢呋喃。600 ml石油醚打浆,过滤干燥得到0.73 mol硫代丁内
酯。
56.步骤二、取0.73 mol硫代丁内酯,7.5 g三溴化磷溶解在300 ml氯仿中,滴加1.387 mol溴素,控温0℃~10℃反应8 h,滴加水500 ml,搅拌分液,氯仿减压浓缩,375 ml石油醚打浆,过滤干燥得到0.63 mol2-溴硫代丁内酯。
57.步骤三、取0.63 mol 2-溴硫代丁内酯溶解在600 ml四氢呋喃中,加入氨基钠0.89 mol控温30℃~40℃反应2 h,降温至10℃,滴加浓盐酸调节ph至5~6,控温低于20℃,降温至10℃析晶2 h,过滤干燥得到dl-高半胱氨酸硫内酯盐酸盐。
58.得到77.80g dl-高半胱氨酸硫内酯盐酸盐,产品收率为60.78%,纯度:≥98.5%。
59.实施例5以实施例1的方法制备获得dl-高半胱氨酸硫内酯盐酸盐。
60.91 g(0.59 mol)dl-高半胱氨酸硫内酯盐酸盐、240 g(2.37 mol)三乙胺溶解在900 ml二氯甲烷中,降温至-10℃~0℃,滴加70 g(0.89 mol)乙酰氯,滴加控温不超过0℃,滴加完毕后缓慢升温至常温反应6 h,300 ml饱和盐水洗涤有机相3次,萃取分层,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,剩余物180 ml乙酸乙酯溶解,5 g活性炭脱色过滤,滴加540 ml石油醚析晶,0℃~10℃析晶4 h,过滤干燥得到83 g(0.52 mol)西替沃酮,产品收率为88.13%,纯度:≥98.0%。
61.西替沃酮氢谱谱图如图1所示,西替沃酮合成路线图如图2所示。
62.以上所述仅为本技术的实施例而已,并不用于限制本技术。对于本领域技术人员来说,本技术可以有各种更改和变化。凡在本技术的精神和原理之内所作的任何修改、等同替换、改进等,均应包含在本技术的权利要求范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1