一种适于工业化生产的三溴新戊醇的制备方法与流程
1.本发明属于化学合成技术领域,具体涉及一种适于工业化生产的三溴新戊醇的制备方法。
背景技术:
2.三溴新戊醇属于脂肪溴,其溴含量73.79%,因其较高的溴含量和结构特殊的稳定性,性质上良好的耐热性、耐水性和光稳定性,常作为阻燃剂使用。三溴新戊醇具有一个可反应的裸露羟基,因此用作高分子量阻燃剂的中间体或反应型阻燃剂。作为中间体与三氯氧磷反应得到添加型高分子量阻燃剂三(三溴新戊基)磷酸酯。同时结合其良好的相容性,能有效与磷结合,应用于聚氨酯的阻燃。与二溴新戊二醇等其他阻燃剂的复配使用,利用两者的可反应型官能团羟基作用,作为优异的反应型阻燃剂。三溴新戊醇在弹性体、涂料和泡沫体的阻燃方面拥有极好的应用前景。此外,在医药技术领域,通过三溴新戊醇制备1,3-环丙二甲醇作为孟鲁司特钠侧链关键中间体,比二溴新戊二醇更具优势。
3.现有较常用的三溴新戊醇合成方法包括:纯气相法,用浓硫酸对氢溴酸进行发气;纯液相法,用氢溴酸长时间回流反应;使用三溴化磷、硫粉和溴素等进行制备。现存方法存在的问题有:纯气相发需要较高的反应条件,高压反应,副产出大量废浓硫酸无法处理,无法实现工业化持续生产的目的,反应控制难,对设备要求和操作高,生产出产品质量无法达到医药级要求,经过溶剂精制,导致收率大大降低,同时存在环保问题;纯液相法因没有高效的催化剂,只能通过高温、加长反应时间来提高三个溴的上溴量,由于高温长时间反应,导致严重的醚化、焦油化副反应,产品色度较高,纯度低,无法达到指标要求,氢溴酸过量量较大,产品的成品高,无法实现工业化生产;三溴化磷法采用价格较高的三溴化磷进行反应,导致生产成本高,同时存在反应过程磷元素无法处理、含磷废水难以处理、废水量大cod高等环保问题,另外三溴化磷的特殊危险性,制备过程存在工艺要求高、设备要求高、操作过程苛刻等安全问题,此法不适应与工业化制备;溴素和硫粉为溴化原料的制备过程,反应效率极低,产品质量差,无法达到需求。
4.如专利cn110903163a提供了一种三溴新戊醇的制备方法,以季戊四醇、溴素和硫粉为原料,有机酸为溶剂加热反应,反应结束后蒸出溶剂和氢溴酸,经甲醇醇解、碳酸钠溶液调ph、烷烃水精制得到成品。该专利采用硫还原会产生二氧化硫废气,硫酸残存在反应体系中加剧副反应的发生,所以收率较低。且产生的硫酸只能靠碳酸钠中和,产生了大量的含盐废水难以处理。
技术实现要素:
5.解决的技术问题:针对上述技术问题,本发明提供了一种适于工业化生产的三溴新戊醇的制备方法,具有收率高、质量好、成本低、反应温和安全、操作简单、生产绿色环保、适合工业化生产的特点,能够达到医药级产品的制备要求。
6.技术方案:一种适于工业化生产的三溴新戊醇的制备方法,包括步骤如下:
s1.溶清:将季戊四醇加入醋酸或甲酸中,再加入酸催化剂,在90-120℃下保温至全部溶清;s2.梯度溴代:s2.1:在90-110℃下向步骤s1所得反应体系中加入氢溴酸,反应后分离出水,得到一溴代体系;s2.2:向一溴代体系中加入氢溴酸,在110-120℃下反应后分离出水,得到二溴代体系;s2.3:向二溴代体系中加入氢溴酸,在120-140℃下反应后分离出水,得到三溴代体系;s3.提纯脱色:s3.1:将三溴代体系蒸干后降温,再加入醇和酸进行酯交换,反应结束后蒸馏分离出醇、酯和水;s3.2:水洗步骤s3.1所得反应体系,再通过碳酸钠溶液调节ph为7.5-8,静置分层;s3.3:向步骤s3.2中分层后的下层料液中加入醇进行结晶,再经干燥后得到白色晶体三溴新戊醇。
7.优选的,所述步骤s2.1中,还向步骤s1所得反应体系中加入抗氧剂。
8.优选的,所述抗氧剂为亚磷酸类抗氧剂。
9.优选的,所述步骤s2.3中,还向二溴代体系中加入酸催化剂。
10.优选的,所述酸催化剂选自硫酸、磷酸、亚磷酸和盐酸中的一种或几种。
11.优选的,所述步骤s2中,在氮气气氛下反应。
12.优选的,所述步骤s2中,通过分水器分离出水或通过负压蒸馏分离出水。
13.优选的,所述分水器包括三通管、冷凝管、回流管和分水管,所述三通管分别连接冷凝管的底端、回流管的顶端和分水管的顶端,所述回流管与分水管之间设有连通管,所述连通管上设有阀门。
14.优选的,所述步骤s3.1中,采用甲醇和乙酸的混合液进行酯交换。
15.优选的,所述步骤s3.3中,还向下层料液中加入活性炭。
16.有益效果:(1)反应所用的物料较少,无危化品,且原料易得,价格低廉;(2)反应条件温和,过程安全可靠,操控简单,安全性高;(3)反应中无大量废气、危废产生,酯分解后通过皂化重新变为醇套用,并副产羧酸钠副产品,调ph产生的废水含盐量极低,ph7.5左右,cod较低,无色无味,可以直接进污水处理系统处理;(4)经工艺优化调整,目标产物纯度在99%以上,收率在96%以上。
17.主要原理如下:整个溴化过程反应氢溴酸的转化率能到98%以上,产品收率99%,得到产品的纯度99%以上,溴代反应过程较温和,无废物产生,较传统工艺无发气过程副产大量废硫酸、无溴化氢气体跑冒滴漏的现象,反应较发气法温和、安全、可控、设备要求低,达到绿色环保工艺要求。
18.梯度溴代前期多羟基化合物不稳定,适于低温反应,温度超过125度,会发生脱水成双键烯烃的反应,导致大量副产物的产生;等上到第三个溴,结构就比较稳定,不会发生脱水反应,同时温度低于130度,上第三个溴的速度就大大降低,所以在反应过程中通过分
水器将水分出体系,然后在上第三个溴时加入酸催化剂如硫酸、磷酸、亚磷酸等,特别是加入亚磷酸能大大加快反应速度,同时产品的色度能低于至5-10hazen。
19.溴代反应前期通入氮气,减少反应体系中氧气,避免体系反应过程中发生醚化反应,同时溴代反应前加入抗氧剂:比如使用亚磷酸类抗氧剂,避免产品高温反应的色变问题,也就是避免焦油化现象的发生,产品加入抗氧剂后色度控制在10 hazen以下。
附图说明
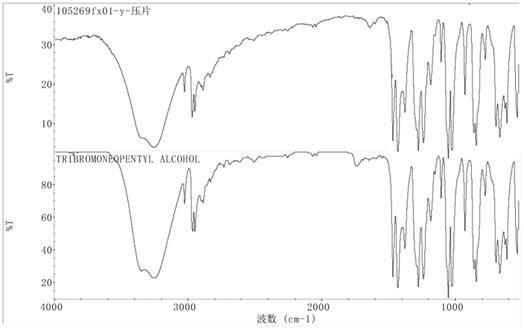
20.图1为实施例1中制得的三溴新戊醇的红外光谱图;图2为实施例1中制得的三溴新戊醇的气象色谱图;图3为实施例3中制得的三溴新戊醇的气象色谱图;图4为分水器的结构示意图。
具体实施方式
21.下面结合附图和具体实施例对本发明作进一步描述。
22.实施例1溶料:将136g季戊四醇、70g醋酸、6g硫酸加入到带有搅拌、温度计、冷凝器的500ml四口烧瓶中,升温到90-120℃,保温直到溶清。硫酸的加入可以加快酯化速度,保证酯化率,减少副反应的发生。
23.梯度溴代:溶清后在90-110℃滴加氢溴酸397.5g,保温2-4h,分水器分水,测定一溴含量80-90%,二溴含量10-15%,三溴含量5-10%;滴加氢溴酸60g,再升温至110-120℃,保温2-6小时,保温完成后,分水器分水,测定二溴含量80-90%,三溴含量10-20%左右;完成后体系中加入10g亚磷酸,滴加氢溴酸53g和30-50g醋酸,再将体系温度升温至120-140度,分水器分水,并保温6-8小时。如图4所示,所述分水器包括三通管、冷凝管、回流管和分水管,所述三通管分别连接冷凝管的底端、回流管的顶端和分水管的顶端,所述回流管与分水管之间设有连通管,所述连通管上设有阀门。回流管中可以乱堆或规整的方式放置填料,回流管上还设有旁通的回流侧管。该分水器从回流管的底部通入溶液升温后的气体,经冷凝管冷凝后,体系中的水落入分水管中,当分水管内出现明显分层后,打开连通管,将分水后的液体回流至回流管中,水从分水管底端排出,分水后的液体由回流管底端回流至体系中。
24.提纯脱色:蒸干后将残余物降温到60℃,加入甲醇180g、硫酸12g,在70℃下酯交换5h,后减压蒸馏,然后向体系中加入水160g,搅拌回流水解4h进行水洗,后升温至80℃,加10wt.%碳酸钠溶液调ph为7.5-8,调好后80℃保温静置40min分层,测定色度为20-50黑增。在下层料液中加入乙醇200g、活性炭2g,脱色1h,后蒸馏回收甲醇。干燥,得到白色晶状成品三溴新戊醇316.1g,收率97.26%,气相纯度99.7%。
25.实施例2将136g季戊四醇、70g醋酸、6g盐酸加入到带有搅拌、温度计、冷凝器的500ml四口烧瓶中,升温到105-110℃,保温直到溶清,溶清后滴加氢溴酸397.5g,保温4h,直到氢溴酸反应完全后,此时三溴类产物达50%以上,降温到85℃,在-0.075mpa真空下加热到110℃减压蒸馏出水;蒸馏结束后加入溶剂醋酸30g,升温至120-135℃,滴加氢溴酸60g,保温3h,直到氢溴酸反应完全后,此时三溴类产物达80%以上,降温到85℃,在-0.075mpa真空下加热到
120℃减压蒸馏出水;蒸馏结束后加入溶剂醋酸20g,升温至125-130℃,滴加氢溴酸53g,保温3h,此时三溴类产物达98%以上,降温到85℃,在-0.075mpa真空下加热到120℃减压蒸馏出水、醋酸以及少量的氢溴酸,再次降温到85℃,在-0.09mpa真空下加热到120℃减压蒸馏出残余水、醋酸以及少量的氢溴酸。蒸干后残余物降温到60℃,加入甲醇180g、盐酸12g,在70℃下酯交换5h,蒸馏后,水洗,加碳酸钠调ph为7.5-8,调好后80℃保温静置40min分层,经甲醇醇结晶、干燥,得到白色晶状成品三溴新戊醇317.1g,收率98.5%,纯度98%(gc)。
26.实施例3将实施例2中的成品加入到甲醇和乙酸质量比为5:1的溶剂200-500g中,在60-80℃回流3-5小时,最后提纯出2分钟和15及18分钟杂质峰,产品气相纯度99.7%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1