一种含氟硅氧烷化合物及其制备方法和应用与流程
1.本发明属于含氟硅氧烷新材料领域,具体涉及一种含氟硅氧烷化合物及其制备方法和应用。
背景技术:
2.随着疏水涂层研究的渗入,人们对涂层的要求也越来越高,根据应用场景的不同合成具有特定功能的涂层已经成为未来疏水行业的趋势,譬如应用在数码产品的触摸屏、半导体或者汽车挡风玻璃等基材表面的防污疏水涂层等。这是因为经常被触摸的玻璃等基材容易被汗渍、污渍污染,而被污染后又不易清洁,从而需要特定的清洗剂清洗,但此过程可能划伤基材表面。
3.目前主要通过有机硅氧烷、丙烯酸酯和异氰酸酯来改善基材表面的抗污性能,其共同点都是以全氟聚醚为主链,如dakin和dow corning公司通过硅氢加成制备了全氟聚醚烷基硅氧烷防指纹液dupont公司和刘亮等人采用全氟聚醚甲酯与氨基硅氧烷,经氨解反应获得了氟硅涂覆剂,3m公司采用端氨基全氟聚醚与3-(甲基丙烯酰氧)丙基三甲氧基硅烷进行michael加成,制备了氟硅表面处理剂,这些有机硅改性的全氟聚醚,不仅具有优异的疏水疏油、耐高低温和自清洁等性能,而且安全可靠、无毒、无污染、无腐蚀,但因耐久性差使得应用领域变窄。
技术实现要素:
4.针对现有技术中存在的问题和不足,本发明的目的在于提供一种含氟硅氧烷化合物及其制备方法和应用。
5.基于上述目的,本发明采用如下技术方案:
6.本发明第一方面提供了一种含氟硅氧烷化合物,其结构式如下所示:
7.[0008][0009]
其中,pfpe为全氟聚醚基团;
[0010]-x表示碳原子数为0-2的直链或支链烷基链;
[0011]-y1、-y2、-y3,均选自-h、-ch3、式(b)所示基团或式(c)所示基团,且-y1、-y2、-y3中至少有一个选自式(b)所示基团或式(c)所示基团;式(b)所示基团、式(c)所示基团中r为-och3、-och2ch3。
[0012]
优选地,所述全氟聚醚基团的分子量为1000~2000。
[0013]
更加优选地,所述全氟聚醚基团的结构式如下所示:
[0014][0015]
其中,n是5~11的整数。
[0016]
本发明第二方面提供了一种上述第一方面所述的含氟硅氧烷化合物的制备方法,包括如下步骤:
[0017]
(1)将含氟稀释剂、氟化盐和甲醇加入反应器中,并将反应器温度升至25~30℃后,加入全氟聚醚,搅拌反应,反应结束后得到混合液a,将混合液a进行后处理得到中间产物ⅰ,所述中间产物ⅰ为全氟聚醚甲酯;
[0018]
(2)将中间产物ⅰ、有机极性溶剂、还原剂加入反应器中,在20~30℃下搅拌进行反应,反应结束后得到混合液b,将混合液b进行后处理得到中间产物ⅱ,所述中间产物ⅱ为全氟聚醚醇;
[0019]
(3)将中间产物ⅱ、缚酸剂加入反应器中,在0~5℃下加入丙烯酰氯,加入完毕后搅拌进行反应,反应结束后得到混合液c,将混合液c进行后处理得到中间产物ⅲ,所述中间产物ⅲ为全氟聚醚丙烯酸酯;
[0020]
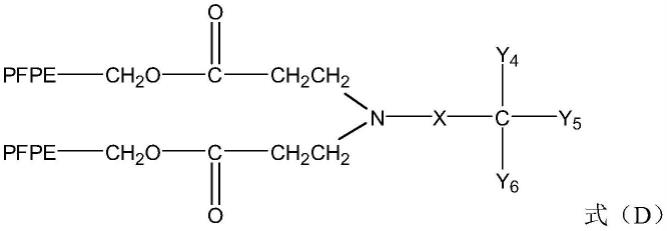
(4)将溶剂、碱性溶液、端基为氨基的醇类化合物加入反应器中,将反应器温度升至40~60℃后,再向反应器中加入中间产物ⅲ,搅拌进行反应,反应结束后得到混合液d,将混合液d进行后处理得到中间产物ⅳ,所述中间产物ⅳ的结构式如式(d)所示:
[0021][0022]
其中,pfpe为全氟聚醚基团;
[0023]-x表示碳原子数为0-2的直链或支链烷基链;
[0024]-y4、-y5、-y6,均选自-h、-ch3、-oh或-ch2oh,且-y4、-y5、-y6中至少有一个选自-oh或-ch2oh;
[0025]
(5)将相转移催化剂、卤代单烯烃、碱液加入反应器中,将反应器温度升至50~70℃后,再向反应器中加入中间产物ⅳ,搅拌并进行反应,反应结束后得到混合液e,将混合液e进行后处理得到中间产物
ⅴ
,所述中间产物
ⅴ
的结构式如式(e)所示:
[0026][0027]
其中,pfpe为全氟聚醚基团;
[0028]-x表示碳原子数为0-2的直链或支链烷基链;
[0029]-y7、-y8、-y9,均选自-h、-ch3、-och2ch=ch2或-ch2och2ch=ch2,且-y7、-y8、-y9中至少有一个选自-och2ch=ch2或-ch2och2ch=ch2;
[0030]
(6)在保护气体氛围下,将中间产物
ⅴ
、催化剂加入反应器中,将反应器温度升至70~90℃后,再向反应器中加入三氯硅烷,搅拌并进行反应,反应结束后得到混合液f,将混合液f进行后处理得到中间产物ⅵ,所述中间产物ⅵ的结构式如式(f)所示:
[0031][0032]
其中,pfpe为全氟聚醚基团;
[0033]-x表示碳原子数为0-2的直链或支链烷基链;
[0034]-y
10
、-y
11
、-y
12
,均选自-h、-ch3、-och2ch2ch2sicl3或-ch2och2ch2ch2sicl3,且-y
10
、-y
11
、-y
12
中至少有一个选自-och2ch2ch2sicl3或-ch2och2ch2ch2sicl3;
[0035]
(7)在保护气体氛围下,将中间产物ⅵ加入反应器中,在0~5℃下加入格氏试剂,
加入完毕后将反应器温度升50~70℃搅拌反应,反应结束后得到混合液g,将混合液g进行后处理得到中间产物ⅶ,所述中间产物ⅶ的结构式如式(g)所示:
[0036][0037]
其中,pfpe为全氟聚醚基团;
[0038]-x表示碳原子数为0-2的直链或支链烷基链;
[0039]-y
13
、-y
14
、-y
15
,均选自-h、-ch3、式(h)所示基团或式(i)所示基团,且-y
13
、-y
14
、-y
15
中至少有一个选自式(h)所示基团或式(i)所示基团;
[0040]
(8)在保护气体氛围下,将中间产物ⅶ、催化剂加入反应器中,将反应器温度升至70~90℃后,再向反应器中加入含有硅氢键的硅烷偶联剂,搅拌并进行反应,反应结束后得到混合液h,将混合液h进行后处理得到最终产物;所述最终产物的结构式如下所示:
[0041][0042]
其中,pfpe为全氟聚醚基团;
[0043]-x表示碳原子数为0-2的直链或支链烷基链;
[0044]-y1、-y2、-y3,均选自-h、-ch3、式(b)所示基团或式(c)所示基团,且-y1、-y2、-y3中至少有一个选自式(b)所示基团或式(c)所示基团;
[0045]
式(b)所示基团、式(c)所示基团中r为-och3、-och2ch3。
[0046]
优选地,步骤(1)中所述全氟聚醚分子量为1000~2000。
[0047]
更加优选地,所述全氟聚醚的结构式如下所示:
[0048][0049]
其中,n是5~11的整数。
[0050]
更加优选地,所述全氟聚醚基团的结构式如下所示:
[0051][0052]
其中,n是5~11的整数。
[0053]
优选地,步骤(1)中所述含氟稀释剂为为三氟三氯乙烷、全氟丁基甲醚、全氟丁基乙醚中的至少一种;所述含氟稀释剂与全氟聚醚的体积比为(1~3)∶1。
[0054]
优选地,步骤(1)中所述氟化盐为氟化钾、氟化钠、氟化铯中的至少一种;所述氟化盐与全氟聚醚的摩尔比为(1~1.2)∶1。
[0055]
优选地,步骤(1)中所述甲醇与全氟聚醚的摩尔比为(10~15)∶1。
[0056]
更加优选地,步骤(1)中全氟聚醚的加入方式为滴加。
[0057]
更加优选地,步骤(1)中所述搅拌反应时间为8h。
[0058]
更加优选地,所述中间产物ⅰ结构式如下所示:
[0059][0060]
其中,pfpe为全氟聚醚基团。
[0061]
优选地,步骤(2)中所述有机极性溶剂为无水乙醇、四氢呋喃中的至少一种;所述有机极性溶剂与中间产物ⅰ的体积比为(1~3)∶1
[0062]
优选地,步骤(2)中所述还原剂为四氢铝锂、硼氢化钠中的至少一种;所述还原剂与中间产物ⅰ的物质的量之比为(0.5~1)∶1。
[0063]
更加优选地,步骤(2)中所述还原剂分批加入反应器中,所述分批加入速率为0.5g/h。
[0064]
更加优选地,步骤(2)中待还原剂完全加入反应器后,所述搅拌反应时间为4~6h。
[0065]
更加优选地,所述中间产物ⅱ的结构式如下所示:
[0066]
pfpe-ch2oh
[0067]
其中,pfpe为全氟聚醚基团。
[0068]
优选地,步骤(3)中所述缚酸剂为三乙胺、吡啶中的至少一种;所述缚酸剂与中间产物ⅱ的物质的量之比为(1.1~1.3)∶1。
[0069]
优选地,步骤(3)中所述丙烯酰氯与中间产物ⅱ的物质的量之比为(1.3~1.5)∶1。
[0070]
更加优选地,步骤(3)中丙烯酰氯的加入方式为滴加。
[0071]
更加优选地,步骤(3)中所述搅拌反应时间为12h。
[0072]
更加优选地,所述中间产物ⅲ的结构式如下所示:
[0073][0074]
其中,pfpe为全氟聚醚基团。
[0075]
优选地,步骤(4)中所述端基为氨基的醇类化合物为端基为氨基的一元醇、端基为氨基的二元醇或端基为氨基的三元醇;所述端基为氨基的一元醇为nh2ch2ch2oh、nh2ch2ch2ch2oh、nh2ch
2-ch(ch3)(oh)、nh2c(ch3)2(ch2oh)中的至少一种;所述端基为氨基的二元醇为nh2ch(ch2oh)2、nh2ch
2-ch(oh)-ch2oh、nh2c(ch3)(ch2oh)2中的至少一种;所述端基为氨基的三元醇为nh2c(ch2oh)3;所述端基为氨基的醇类化合物与中间产物ⅲ的摩尔比为0.5∶1。
[0076]
优选地,步骤(4)中所述碱性溶液为质量分数为40wt%~60wt%的碳酸钠或碳酸钾的水溶液;所述碱性溶液中溶质与中间产物ⅲ的物质的量之比为(0.02~0.05)∶1
[0077]
优选地,步骤(4)中所述溶剂为异丙醇;所述溶剂与中间产物ⅲ的体积比为(1~2)∶1。
[0078]
更加优选地,步骤(4)中中间产物ⅲ的加入方式为滴加。
[0079]
更加优选地,步骤(4)中所述搅拌反应时间为12~16h。
[0080]
优选地,步骤(5)中所述相转移催化剂为四丁基溴化铵、四丁基碘化铵、四甲基溴化铵中的至少一种;所述相转移催化剂与中间产物ⅳ的物质的量之比为1∶(3~10)。
[0081]
更加优选地,当步骤(4)中端基为氨基的醇类化合物为端基为氨基的一元醇或端基为氨基的二元醇时,步骤(5)中所述相转移催化剂与中间产物ⅳ的物质的量之比为1∶(4.5~10)。
[0082]
优选地,步骤(5)中所述卤代单烯烃为溴丙烯、氯丙烯中的至少一种;所述卤代单烯烃与中间产物ⅳ的物质的量之比为(2~9)∶1。
[0083]
更加优选地,当步骤(4)中端基为氨基的醇类化合物为端基为氨基的一元醇或端基为氨基的二元醇时,步骤(5)中所述卤代单烯烃与中间产物ⅳ的物质的量之比为(2~6)∶1。
[0084]
优选地,步骤(5)中所述碱液为质量分数为30wt%~50wt%的氢氧化钠或氢氧化钾的水溶液;所述碱液中溶质与中间产物ⅳ的物质的量之比为(1~3.3)∶1。
[0085]
更加优选地,当步骤(4)中端基为氨基的醇类化合物为端基为氨基的一元醇或端基为氨基的二元醇时,步骤(5)中所述碱液中溶质与中间产物ⅳ的物质的量之比为(1~2.2)∶1。
[0086]
更加优选地,步骤(5)中中间产物ⅳ的加入方式为滴加。
[0087]
更加优选地,步骤(5)中所述搅拌反应时间为12~24h。
[0088]
优选地,步骤(6)中所述催化剂为氯铂酸的异丙醇溶液,所述氯铂酸的异丙醇溶液的浓度为0.05mol/l;所述氯铂酸与中间产物
ⅴ
的物质的量之比为1∶(3000~30000)。
[0089]
更加优选地,当步骤(4)中端基为氨基的醇类化合物为端基为氨基的一元醇或端基为氨基的二元醇时,步骤(6)中所述氯铂酸与中间产物
ⅴ
的物质的量之比为1∶(5000~30000)。
[0090]
优选地,步骤(6)中所述三氯硅烷与中间产物
ⅴ
的物质的量之比为(1~6)∶1。
[0091]
更加优选地,当步骤(4)中端基为氨基的醇类化合物为端基为氨基的一元醇或端基为氨基的二元醇时,步骤(6)中所述三氯硅烷与中间产物
ⅴ
的物质的量之比为(1~4)∶1。
[0092]
更加优选地,步骤(6)中所述搅拌反应时间为8~12h。
[0093]
更加优选地,所述步骤(6)中三氯硅烷的加入方式为滴加;将反应器温度升至70~90℃保温加热搅拌1h再滴加三氯硅烷。
[0094]
优选地,步骤(7)中所述格氏试剂为烯丙基氯化镁或烯丙基溴化镁的四氢呋喃溶液;所述烯丙基氯化镁或烯丙基溴化镁的四氢呋喃溶液的浓度为2mol/l;所述格氏试剂与中间产物ⅵ的物质的量之比为(3~10.5)∶1。
[0095]
更加优选地,当步骤(4)中端基为氨基的醇类化合物为端基为氨基的一元醇或端基为氨基的二元醇时,步骤(7)中所述格氏试剂与中间产物ⅵ的物质的量之比为(3~7)∶1。
[0096]
更加优选地,步骤(7)中格氏试剂的加入方式为滴加。
[0097]
更加优选地,步骤(7)中所述搅拌反应时间为12~24h。
[0098]
优选地,步骤(8)中所述催化剂为氯铂酸的异丙醇溶液,所述氯铂酸的异丙醇溶液的浓度为0.05mol/l;所述氯铂酸与中间产物ⅶ的物质的量之比为1∶(1000~10000)。
[0099]
更加优选地,当步骤(4)中端基为氨基的醇类化合物为端基为氨基的一元醇或端基为氨基的二元醇时,步骤(8)中所述氯铂酸与中间产物ⅶ的物质的量之比为1∶(2000~10000)。
[0100]
优选地,步骤(8)中所述含硅氢键的硅烷偶联剂为三甲氧基硅烷、三乙氧基硅烷中的至少一种;所述含硅氢键的硅烷偶联剂与中间产物ⅶ的物质的量之比为(3~13.5)∶1。
[0101]
更加优选地,当步骤(4)中端基为氨基的醇类化合物为端基为氨基的一元醇或端基为氨基的二元醇时,步骤(8)中所述含硅氢键的硅烷偶联剂与中间产物ⅶ的物质的量之比为(3~9)∶1。
[0102]
更加优选地,步骤(8)中含有硅氢键的硅烷偶联剂的加入方式为滴加。
[0103]
更加优选地,步骤(8)中所述搅拌反应时间为24~36h。
[0104]
更加优选地,所述保护气体为氮气。
[0105]
更加优选地,步骤(1)中所述混合液a的后处理方式为:向混合液a中加水至中性,静置后收集下层有机相,减压除去含氟稀释剂,再加入甲醇,充分萃取后静置,收集下层有机相,减压蒸除甲醇。
[0106]
更加优选地,步骤(2)中所述混合液b的后处理方式为:将混合液b水洗2~3次,静置后收集下层有机相,再加入甲醇,充分萃取后静置,收集下层有机相,减压蒸除甲醇。
[0107]
更加优选的,步骤(3)中所述混合液c的后处理方式为:将混合液c以无水乙醇萃取3次,每次充分萃取后静置,收集下层有机相,最后减压蒸除乙醇。
[0108]
更加优选地,步骤(4)中所述混合液d的后处理方式为:将混合液d水洗至中性,静置后收集下层有机相,再加入甲醇,充分萃取后静置,收集下层有机相,减压蒸除甲醇。
[0109]
更加优选地,步骤(5)中所述混合液e的后处理方式为:将混合液e水洗2次,静置后收集下层有机相,再加入甲醇,充分萃取后静置,收集下层有机相,减压蒸除甲醇。
[0110]
更加优选地,步骤(6)中所述混合液f的后处理方式为:减压蒸除混合液f中残留的三氯硅烷。
[0111]
更加优选地,步骤(7)中所述混合液g的后处理方式为:将混合液g过滤后收集滤液,蒸除滤液中的四氢呋喃,再加入少量去离子水清洗三次,静置后收集下层有机相,再加入甲醇,充分萃取后静置,收集下层有机相,减压蒸除甲醇,再加入含氟稀释剂过滤,蒸除滤液中残留含氟稀释剂。
[0112]
更加优选地,步骤(8)中所述混合液h的后处理方式为:将水分小于70ppm的无水乙醇加入混合液h中萃取残余的硅烷偶联剂,充分萃取后静置,收集下层有机相,减压蒸除残留乙醇。
[0113]
本发明第三方面提供了如上述第一方面所述的含氟硅氧烷化合物在防指纹涂层中的应用,尤其是半导体、手机3c产业、钢化玻璃、汽车玻璃、钟表等行业,优选在半导体硅片表面的涂层产品。
[0114]
本发明第四方面提供了一种防指纹涂层的制备方法,包括如下步骤:将上述第一方面所述的含氟硅氧烷化合物用稀释剂稀释,得到稀释溶液,然后向稀释溶液中加入表面处理剂并搅拌均匀,得到所述防指纹涂料溶液,将所述涂料溶液涂覆在基材上,在110~150℃温度下固化成膜,得到所述防指纹涂层。
[0115]
优选地,所述稀释剂为全氟丁基甲醚、全氟丁基乙醚中的至少一种;所述表面处理
剂为氨丙基三甲氧基硅烷、氨丙基三乙氧基硅烷中的至少一种。
[0116]
更加优选地,所述含氟硅氧烷化合物用稀释剂稀释至0.2~0.5wt%;所述表面处理剂的加入量为0.1~0.3wt%。
[0117]
与现有技术相比,本发明的有益效果如下:
[0118]
(1)本发明制备的含氟硅氧烷化合物的分子链中一方面具有极多的水解集团,水解后便于与基材表面形成多个化学键,形成交联结构,以增加固化涂层的硬度,进而增加该涂层的使用寿命且可以有效保护基材,能大大提高耐磨性能与使用寿命;另一方面分子链具有2条超疏水的全氟聚醚链,在制备的防指纹涂层的固化过程中,全氟聚醚结构会由于表面能的作用向涂层表面迁移聚集,使形成的涂层具有优异的疏水疏油性能以及优异的防污性能。
[0119]
(2)本发明制备的超疏水型防指纹涂层中引入硅烷偶联剂使得有机物与基材牢牢粘连,具有耐摩擦、耐久性能优异等特点,可被应用于手机触摸屏及高端数码产品、半导体等行业。
[0120]
(3)此外,较现有制备工艺,本发明采用低成本、绿色的合成路线及后处理工艺,获得转化率、收率较高的树枝状多官能团结构的全氟聚醚硅氧烷。
具体实施方式
[0121]
为使本发明的目的、技术方案及优点更加清楚明白,以下通过实施例对本发明作进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
[0122]
需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互组合。下面将结合实施例来详细说明本技术。
[0123]
实施例1:制备含氟硅氧烷化合物
[0124]
本实施例提供一种含氟硅氧烷化合物,其制备方法包括如下步骤:
[0125]
(1)向反应器中加入110~150ml全氟丁基甲醚、3.2~6.4g氟化钾及23g~34g甲醇,将反应器温度升至30℃,然后缓慢滴加100g k型全氟聚醚(分子量1000~2000),滴加完毕后继续搅拌反应8h,反应完毕后得到混合液a;向混合液a中加入大量水洗将有机相洗至中性,静置后收集下层有机相,蒸除下层有机相中的全氟丁基甲醚,再向蒸馏残液中加入甲醇,萃取其中残留水分,充分萃取后静置,收集下层有机相,最后减压除去甲醇,得到99.8~100.2g中间产物ⅰ。所述中间产物ⅰ为全氟聚醚甲酯,结构式如下所示:
[0126][0127]
其中,pfpe为全氟聚醚基团,pfpe分子量为1000~2000。
[0128]
所述k型全氟聚醚的结构式如下所示:
[0129]
[0130]
其中,n是5~11的整数。
[0131]
(2)向反应器中加入100g中间产物ⅰ,再向反应器中加入110~150ml无水乙醇,然后再每隔1h加入0.5g硼氢化钠,共计加入硼氢化钠1.7~1.9g,控制反应温度在25℃,待硼氢化钠加入完毕后再次搅拌反应6h,反应结束后得到混合液b;将混合液b用大量水洗至中性,静置后收集下层有机相,然后向有机相中加入甲醇萃取残留水分,充分萃取后静置,收集下层有机相,减压蒸除甲醇得到96.6~97.1g中间产物ⅱ。所述中间产物ⅱ为全氟聚醚醇,结构式如下所示:
[0132]
pfpe-ch2oh
[0133]
其中,pfpe为全氟聚醚基团,pfpe分子量为1000~2000。
[0134]
(3)将100g中间产物ⅱ置于反应器中,再向反应器中加入6.6~12.3g三乙胺,将体系降温至0℃,再向反应器中缓慢滴加6.8~12.9g丙烯酰氯,滴加结束后搅拌进行反应,反应结束后得到混合液c;降温至室温,向混合液c中加入大量乙醇萃取3次,每次充分萃取后静置,收集下层有机相,最后减压蒸除乙醇得到100.7~103g中间产物ⅲ。所述中间产物ⅲ为全氟聚醚丙烯酸酯,结构式如下所示:
[0135][0136]
其中,pfpe为全氟聚醚基团,pfpe分子量为1000~2000。
[0137]
(4)将93~110ml异丙醇、质量分数为50wt%的碳酸钾水溶液(其中,碳酸钾的质量为0.2~0.4g)、1.5~2.8g乙醇胺加入反应器中,将反应器温度升至50℃,待温度稳定后向反应器中缓慢滴加100g中间产物ⅲ,滴加完毕后继续搅拌反应12h,反应结束后得到混合液d;降温至室温,向混合液d中加入去离子水将有机相洗至中性,静置后收集下层有机相,再向有机相中加入甲醇萃取残留水分,充分萃取后静置,收集下层有机相,减压蒸除甲醇得到100.5~100.9g中间产物ⅳ。所述中间产物ⅳ的结构式如下所示:
[0138][0139]
其中,pfpe为全氟聚醚基团,pfpe分子量为1000~2000。
[0140]
(5)向反应器中加入0.78~1.68g四丁基溴化铵,8.8~12.6g溴丙烯,质量分数为30wt%的氢氧化钠水溶液(其中,氢氧化钠的质量为0.97~1.89g)缓慢升温至60℃,再向反应器中缓慢滴加100g中间产物ⅳ,继续搅拌反应12h,反应结束后得到混合液e;降温至室温,向混合液e中加入大量去离子水将有机相洗至中性,静置后收集下层有机相,再向有机相中加入甲醇萃取残留水分,充分萃取后静置,收集下层有机相,减压除去甲醇得到100.1~100.6g中间产物
ⅴ
。所述中间产物
ⅴ
的结构式如下所示:
[0141][0142]
其中,pfpe为全氟聚醚基团,pfpe分子量为1000~2000。
[0143]
(6)在n2保护的氛围下,将100g中间产物
ⅴ
置于反应器中,再向其中加入0.04~0.09ml氯铂酸的异丙醇溶液,搅拌并缓慢升温至80℃,待温度稳定后缓慢滴加5~9.5g三氯硅烷,滴加完毕后继续搅拌反应8h,得到混合液f;减压除去混合液f中未反应完的三氯硅烷,得到97.4~100g中间产物ⅵ。所述中间产物ⅵ的结构式如下所示:
[0144][0145]
其中,pfpe为全氟聚醚基团,pfpe分子量为1000~2000。
[0146]
(7)在n2保护的氛围下,先将100g中间产物ⅵ加入反应器中,将反应器降温至0℃,再向其中缓慢滴加40.7~72ml烯丙基氯化镁的四氢呋喃溶液,滴加完毕后缓慢升温至60℃,继续搅拌反应12h,反应结束后得到混合液g;降温至室温,先将混合液g进行过滤后收集滤液,然后蒸除滤液中的四氢呋喃,再向蒸馏残液中加入少量去离子水清洗三次,静置后收集下层有机相,再向下层有机相中加入甲醇萃取残留水分,充分萃取后静置,收集下层有机相,减压除去甲醇得到99.5~100.3g中间产物ⅶ。所述中间产物ⅶ的结构式如下所示:
[0147][0148]
其中,pfpe为全氟聚醚基团,pfpe分子量为1000~2000。
[0149]
(8)在n2保护的氛围下,将100g中间产物ⅶ和0.09~0.17ml氯铂酸的异丙醇溶液加入反应器中,搅拌加热至80℃,待温度稳定后,缓慢滴加11~20.6g三甲氧基硅烷,滴加完毕后继续搅拌反应24h,反应结束后得到混合液h;降温至室温,向混合液h中加入少量水分小于70ppm的无水乙醇,萃取未反应完的三甲氧基硅烷,充分萃取后静置,收集下层有机相,减压蒸除乙醇,得到98.9~105g最终产物。所述最终产物的结构式如下所示:
[0150][0151]
其中,pfpe为全氟聚醚基团,pfpe分子量为1000~2000;r为-och3。
[0152]
实施例1-1~1-3的区别在于,各步骤的工艺参数不同,具体如表1所示。
[0153]
表1实施例1-1~1-3中的工艺参数值
[0154][0155][0156]
实施例2:制备含氟硅氧烷化合物
[0157]
本实施例提供一种含氟硅氧烷化合物,其制备方法包括如下步骤:
[0158]
(1)向反应器中加入150ml全氟丁基甲醚、3.2g氟化钾及23g甲醇,将反应器温度升至30℃,然后缓慢滴加100g k型全氟聚醚(分子量2000),滴加完毕后继续搅拌反应8h,反应完毕后得到混合液a;向混合液a中加入大量水洗将有机相洗至中性,静置后收集下层有机相,蒸除下层有机相中的全氟丁基甲醚,再向蒸馏残液中加入甲醇,萃取其中残留水分,充
分萃取后静置,收集下层有机相,最后减压除去甲醇,得到99.8g中间产物ⅰ。所述中间产物ⅰ为全氟聚醚甲酯,结构式如下所示:
[0159][0160]
其中,pfpe为全氟聚醚基团,pfpe分子量约为2000。
[0161]
所述k型全氟聚醚的结构式如下所示:
[0162][0163]
(2)向反应器中加入100g中间产物ⅰ,再向反应器中加入150ml无水乙醇,然后再每隔1h加入0.5g硼氢化钠,共计加入硼氢化钠1.7g,控制反应温度在25℃,待硼氢化钠加入完毕后再次搅拌反应6h,反应结束后得到混合液b;将混合液b用大量水洗至中性,静置后收集下层有机相,然后向有机相中加入甲醇萃取残留水分,充分萃取后静置,收集下层有机相,减压蒸除甲醇得到97.1g中间产物ⅱ。所述中间产物ⅱ为全氟聚醚醇,结构式如下所示:
[0164]
pfpe-ch2oh
[0165]
其中,pfpe为全氟聚醚基团,pfpe分子量约为2000。
[0166]
(3)将100g中间产物ⅱ置于反应器中,再向反应器中加入6.6g三乙胺,将体系降温至0℃,再向反应器中缓慢滴加6.8g丙烯酰氯,滴加结束后搅拌进行反应,反应结束后得到混合液c;降温至室温,向混合液c中加入大量乙醇萃取3次,每次充分萃取后静置,收集下层有机相,最后减压蒸除乙醇得到100.7g中间产物ⅲ。所述中间产物ⅲ为全氟聚醚丙烯酸酯,结构式如下所示:
[0167][0168]
其中,pfpe为全氟聚醚基团,pfpe分子量约为2000。
[0169]
(4)将75ml异丙醇、质量分数为50wt%的碳酸钾水溶液(其中,碳酸钾的质量为0.33g)、2.3g2-氨基-1,3-丙二醇加入反应器中,将反应器温度升至50℃,待温度稳定后向反应器中缓慢滴加100g中间产物ⅲ,滴加完毕后继续搅拌反应16h,反应结束后得到混合液d;降温至室温,向混合液d中加入去离子水将有机相洗至中性,静置后收集下层有机相,再向有机相中加入甲醇萃取残留水分,充分萃取后静置,收集下层有机相,减压蒸除甲醇得到94.5g中间产物ⅳ。所述中间产物ⅳ的结构式如下所示:
[0170][0171]
其中,pfpe为全氟聚醚基团,pfpe分子量约为2000。
[0172]
(5)向反应器中加入1.58g四丁基溴化铵,12.3g溴丙烯,质量分数为30wt%的氢氧化钠水溶液(其中,氢氧化钠的质量为1.87g),缓慢升温至70℃,再向反应器中缓慢滴加100g中间产物ⅳ,继续搅拌反应24h,反应结束后得到混合液e;降温至室温,向混合液e中加入大量去离子水将有机相洗至中性,静置后收集下层有机相,再向有机相中加入甲醇萃取残留水分,充分萃取后静置,收集下层有机相,减压除去甲醇得到96.8g中间产物
ⅴ
。所述中间产物
ⅴ
的结构式如下所示:
[0173][0174]
其中,pfpe为全氟聚醚基团,pfpe分子量约为2000。
[0175]
(6)在n2保护的氛围下,将100g中间产物
ⅴ
置于反应器中,再向其中加入0.08ml氯铂酸的异丙醇溶液,搅拌并缓慢升温至80℃,待温度稳定后缓慢滴加9.4g三氯硅烷,滴加完毕后继续搅拌反应12h,得到混合液f;减压除去混合液f中未反应完的三氯硅烷,得到93g中间产物ⅵ。所述中间产物ⅵ的结构式如下所示:
[0176][0177]
其中,pfpe为全氟聚醚基团,pfpe分子量约为2000。
[0178]
(7)在n2保护的氛围下,先将100g中间产物ⅵ加入反应器中,将反应器降温至0℃,再向其中缓慢滴加71.2ml烯丙基氯化镁的四氢呋喃溶液,滴加完毕后缓慢升温至60℃,继续搅拌反应24h,反应结束后得到混合液g;降温至室温,先将混合液g进行过滤后收集滤液,然后蒸除滤液中的四氢呋喃,再向蒸馏残液中加入少量去离子水清洗三次,静置后收集下层有机相,再向下层有机相中加入甲醇萃取残留水分,充分萃取后静置,收集下层有机相,减压除去甲醇得到99.5g中间产物ⅶ。所述中间产物ⅶ的结构式如下所示:
[0179][0180]
其中,pfpe为全氟聚醚基团,pfpe分子量约为2000。
[0181]
(8)在n2保护的氛围下,将100g中间产物ⅶ和0.12ml氯铂酸的异丙醇溶液加入反应器中,搅拌加热至80℃,待温度稳定后,缓慢滴加21.6g三甲氧基硅烷,滴加完毕后继续搅拌反应36h,反应结束后得到混合液h;降温至室温,向混合液h中加入少量水分小于70ppm的无水乙醇,萃取未反应完的三甲氧基硅烷,充分萃取后静置,收集下层有机相,减压蒸除乙醇,得到95g最终产物。所述最终产物的结构式如下所示:
[0182][0183]
其中,pfpe为全氟聚醚基团,pfpe分子量约为2000。
[0184]
实施例3
[0185]
本发明实施例提供一种防指纹涂层,其制备方法包括如下步骤:
[0186]
(a)将实施例1-1制备的含氟硅氧烷化合物先以全氟丁基甲醚稀释至0.5wt%,再加入0.3wt%的氨丙基三乙氧基硅烷,得到涂料溶液。
[0187]
(b)手机触摸屏在室温下用丙酮、乙醇分别超声清洗30min,再以去离子水超声清洗30min,得到预处理手机触摸屏;将预处理手机触摸屏用食人鱼溶液(浓硫酸∶双氧水体积比7∶3)中浸泡30min,再以去离子水清洗触摸屏至中性,得到活化手机触摸屏。
[0188]
(c)将步骤(a)中制备的涂料溶液用匀胶机均匀旋涂于活化手机触摸屏上,常温静置30min后,在真空烘箱中以150℃烘烤1h,然后静置于常温、空气湿度为40%的环境中24h,得到所述高耐磨型防指纹涂层。
[0189]
实施例4
[0190]
一种防指纹涂层的制备方法内容与实施例3的基本相同,其不同之处在于:所述含氟硅氧烷化合物为实施例1-2制备的含氟硅氧烷材料。
[0191]
实施例5
[0192]
一种防指纹涂层的制备方法内容与实施例3的基本相同,其不同之处在于:所述含氟硅氧烷材料为实施例1-3制备的含氟硅氧烷材料。
[0193]
实施例6
[0194]
一种防指纹涂层的制备方法内容与实施例3的基本相同,其不同之处在于:步骤(a)中用全氟丁基乙醚稀释至0.2wt%,再加入0.1wt%的氨丙基三乙氧基硅烷;步骤(c)中
在真空烘箱中以110℃烘烤。
[0195]
对比例1
[0196]
一种防指纹涂层的制备方法内容与实施例3的基本相同,其不同之处在于:所述含氟硅氧烷化合物为一种含全氟聚醚硅烷化合物(d1),所述全氟聚醚硅氧烷化合物(d1)结构式如下所示:
[0197][0198]
所述d1的制备方法具体为:以端甲酯化全氟聚醚(分子量3000)为起始原料,与3-氨丙基三甲氧基硅烷合成酰胺键为间隔基的全氟聚醚硅烷。
[0199]
对比例2
[0200]
一种防指纹涂层的制备方法内容与实施例3的基本相同,其不同之处在于:所述含氟硅氧烷化合物为一种氟硅烷化合物(d2),所述氟硅烷化合物(d2)结构式如下所示:
[0201][0202]
其中,p1∶q1=47∶53,且p1+q1≈43。
[0203]
所述d2的制备方法具体为:以全氟聚醚酰氟(分子量2000)为起点,经由格氏试剂反应、三甲氧基硅烷硅氢加成制得。
[0204]
对比例3
[0205]
一种防指纹涂层的制备方法内容与实施例3的基本相同,其不同之处在于:所述含氟硅氧烷化合物为一种含全氟聚醚硅烷化合物(d3),市面现有产品双端硅乙氧基全氟聚醚(分子量2000),所述含全氟聚醚硅烷化合物(d3)结构式如下所示:
[0206][0207]
性能测试:
[0208]
为了探讨本发明制备的防指纹涂层的疏水疏油性、耐污能力、以及耐摩擦能力,发明人分别做了以下实验,即实施例3、实施例4、实施例5、对比例1、对比例2、对比例3,然后将得到的防指纹涂层浸泡于全氟丁基甲醚溶液中,常温下超声10min,再进行接触角测试、防污性能测试、耐摩擦测试。结果如表2所示。测试过程的具体步骤如下所示:
[0209]
1、接触角测试
[0210]
接触角测试采用液滴法,通过玻璃表面的去离子水接触角与正十六烷接触角表征防指纹涂层的疏水疏油性。测试采用jgw-360a水接触角测试仪测量水和正十六烷的接触角,并在室温下进行测量。将待测手机触屏玻璃样品平铺在接触角测定仪的水平平台上并
固定,且液滴尺寸为8微升,同一样品测5个点,取其平均值。接触角越大,表面能越小。
[0211]
2、防污性能测试
[0212]
油性笔测试:使用市售的油基油墨笔在手机玻璃屏的固化薄膜表面上绘制蓝色线。根据蓝色油墨收缩情况评价其耐污能力高低。评判标准如下:
[0213]
c——不收缩,成线;
[0214]
b——收缩成虚线;
[0215]
a——收缩成点。
[0216]
指纹测试:涂层粘上指纹,通过用kimwipes无尘布在固化膜表面反复擦拭5次,形成的指纹去除情况。目测评价其指纹去除的简易性。
[0217]
c——擦拭后,明显留下污点;
[0218]
b——擦拭后,去除大部分污点,有微痕;
[0219]
a——擦拭后,污点全部去除。
[0220]
3、耐摩擦测试
[0221]
用钢丝绒(bonstar#0000,20mm直径)负载1kg压力,以60次/分钟的频率水平在涂层样板上反复摩擦3000次后,测试水接触角。
[0222]
表2本发明制备的高耐磨型防指纹涂层的抗指纹性能测试结果
[0223]
测试项目实施例3实施例4实施例5对比例1对比例2对比例3水接触角(
°
)123130144107108112正十六烷接触角(
°
)727577707170油性笔测试aaabaa指纹测试aaabaa耐摩擦测试(
°
)12012513795100106
[0224]
从表2可以看出,本发明制备的防指纹层(实施例3~5)经耐摩擦测试前后,水滴在所述防指纹涂层上形成的水滴角的角度变化不大,由此可充分证明所述防指纹涂层与手机触屏玻璃样品之间具有良好的结合力,皆具有良好的抗指纹性及耐摩擦性,较同类抗指纹涂层疏水性及耐磨擦性能有显著提升。同时,由于本发明的制备的防指纹涂层在合成过程中使用低分子量的全氟聚醚,能在降低全氟聚醚硅氧烷涂层产品生产成本的基础上,表现出更加优异的防油污、抗划伤及耐久性等性能,且手感极佳,因此极具实用价值。本发明的防指纹涂层可应用于半导体、手机3c产业、钢化玻璃、汽车玻璃、钟表等行业,尤其是半导体硅片表面。
[0225]
综上所述,本发明有效克服了现有技术中的不足,且具高度产业利用价值。上述实施例的作用在于说明本发明的实质性内容,但并不以此限定本发明的保护范围。本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1