一种1-烷氧基异喹啉化合物的绿色合成方法
1.本发明属于化合物合成技术领域,涉及一种1-烷氧基异喹啉化合物的绿色合成方法。
背景技术:
2.异喹啉结构是重要的有机组成结构,含有该结构片段的化合物大多具有优良的生物和药理活性,如抗真菌、抗肿瘤、抗病毒活性等,因而在医药及其它相关领域有着广泛的应用。其中,1-烷氧基异喹啉化合物具有较好的临床应用。例如,奎尼卡因,可用于表面麻醉,其麻醉性比可卡因强1000余倍,且毒性较低。因而,近年来国内外对含1-烷氧基异喹啉化合物合成研究日益增多。
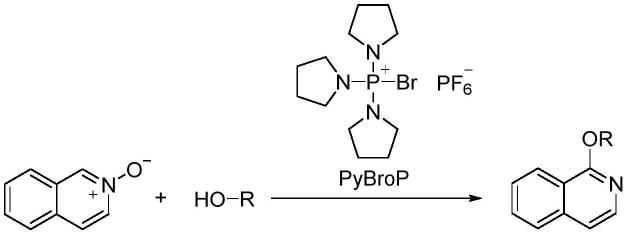
3.氮杂环醚的传统合成主要通过氮杂环卤代物在强碱作用下与醇发生反应,然而此类反应的温度较高,收率低。londregan等人报道了异喹啉的n氧化物为原料,以三吡咯烷基溴化鏻六氟磷酸盐(pybrop)为促进剂,在碱性条件下实现了1-烷氧基异喹啉化合物的合成(org.lett.,2016,18:1362
–
1365)。该方法需要用到较为昂贵的三吡咯烷基溴化鏻六氟磷酸盐(aldrich,280元/克),且含有取代基的异喹啉n氧化物原料需要多步合成。
[0004][0005]
近年来,通过“一锅法”的分子内c-h活化/环化得到了研究人员的广泛重视。例如,hua ruimao等人以二苯炔、腈和醇为原料,在铑催化剂作用下,实现了1-烷氧基-3,4-二芳基取代的异喹啉化合物(j.org.chem.,2021,86,8862-8872)。该反应需要使用昂贵的铑催化剂,且炔烃需要为对称结构。
[0006][0007]
另外,上述方法报道的1-烷氧基异喹啉化合物的3,4-号位置都有相应的取代基团,而3,4号位没有取代基团的1-烷氧基异喹啉化合物的通用合成方法鲜有报道。现有技术中1-烷氧基异喹啉化合物的合成方法通常需要使用有毒易挥发的有机溶剂作为反应介质,且使用的催化剂价格昂贵。有机溶剂、催化剂和添加剂难以回收,不仅造成环境污染,还导致反应成本高。鉴于3,4号位没有被取代的1-烷氧基异喹啉化合物可经由其它反应引入多样性官能团化的重要特性,因此,开发一种原料简单、反应活性高、通用性好、反应成本低、
环境友好的3,4号位没有取代基的1-烷氧基异喹啉化合物绿色合成方法十分必要。
技术实现要素:
[0008]
针对现有技术中的1-烷氧基异喹啉化合物合成方法存在的缺陷,本发明的目的在于,提供一种以苯甲腈类化合物和醇为反应的起始原料,以低共熔物为溶剂,发生加成反应,继而在钴催化剂和添加剂作用下与碳酸亚乙烯酯发生分子内c-h活化/环化反应,制备得到1-烷氧基异喹啉化合物的绿色合成方法,该方法目标产物收率高,原料成本低,环境友好,有利于工业化生产应用。
[0009]
为实现上述目的,本发明采用技术方案是:
[0010]
一种1-烷氧基异喹啉化合物的绿色合成方法,以苯甲腈类化合物和醇为反应的起始原料,以低共熔物为溶剂,发生加成反应,继而在钴催化剂和添加剂作用下与碳酸亚乙烯酯发生分子内c-h活化/环化反应,制备得到1-烷氧基异喹啉化合物;
[0011]
反应路线如下:
[0012][0013]
本发明的1-烷氧基异喹啉化合物中,r1可以为氢、甲基、甲氧基、卤素、氰基、酯基、三氟甲基、硝基等。取代基的位置不限,可以为邻、间、对位。
[0014]
进一步的,r2为c1~c4的直链或支链烷基、三氟乙基、烯丙基等。1-烷氧基异喹啉的3,4号位无取代基。r1和r2取代基类型、位置的选择对目标产物的收率影响并不很明显,实验表明各类取代基基本都能保证目标产物收率在80%以上。
[0015]
作为一个优选的方案,低共熔溶剂选自氯化胆碱/乙酸、氯化胆碱/丙二酸、氯化胆碱/甘油、甜菜碱/六氟异丙醇中的任意一种。优选的氯化胆碱/乙酸低共熔溶剂由氯化胆碱和乙酸按照摩尔比1:1~3组成。优选的氯化胆碱/丙二酸低共熔溶剂由氯化胆碱和丙二酸按照摩尔比1:1~3组成。优选的氯化胆碱/甘油低共熔溶剂由氯化胆碱和甘油按照摩尔比1:1~3组成。优选的甜菜碱/六氟异丙醇低共熔溶剂由甜菜碱和六氟异丙醇按照摩尔比1:1~3组成。在这几类低共熔溶剂中1-烷氧基异喹啉化合物都能顺利生成,目标产物的收率都在60%以上,而甜菜碱/六氟异丙醇是该反应最佳的低共熔溶剂,特别是甜菜碱/六氟异丙醇两者按照摩尔比1:2组成时,获得的目标产物的收率达到最佳。本发明采用的低共熔溶剂使用量按照常规溶剂的添加量,如1-烷氧基异喹啉在低共熔溶剂中的浓度为0.05~0.2mol/l,优选为0.05~0.1mol/l。
[0016]
进一步的,所述的钴催化剂为醋酸钴、cp*co(co)i2和[cocp*(ch3cn)3](sbf6)2中的一种。进一步优选的技术方案还有,所述的钴盐催化剂的用量至少为苯甲腈摩尔投料量的2.5%。钴盐催化剂的最佳用量为5%当量,增加钴盐催化剂用量时,目标产物的收率无明显增加;降低钴盐催化剂用量时,目标产物的收率降低明显;更进一步优选2.5~10%。
[0017]
所述的添加剂用作氧化剂,为醋酸铜、氧化铜、醋酸银、碳酸银中的一种。进一步优选的技术方案还有,添加剂用量为苯甲腈的10~30mol%。添加剂的最佳用量为20mol%。
[0018]
所述的一种1-烷氧基异喹啉化合物的绿色合成方法,所述的苯甲腈:醇:碳酸亚乙烯酯摩尔比为1:10~20:3~5。
[0019]
进一步优选的技术方案还有,苯甲腈:醇:碳酸亚乙烯酯摩尔比为1:20:3。
[0020]
所述的分子内c-h活化/环化反应条件为:在90~120℃下反应12~24h。温度最优选在110℃左右,时间最优为12h,温度进一步升高或延长反应时间,目标产物的收率无增加。
[0021]
所述的分子内c-h活化/环化反应完成后,采用有机溶剂萃取,分离1-烷氧基异喹啉化合物,萃余液为低共熔溶剂、钴盐催化剂和添加剂,可重复使用。有机溶剂如乙酸乙酯等。
[0022]
本发明的优点和有益效果在于:
[0023]
1、本发明提供了一种通用的1-烷氧基异喹啉化合物的绿色合成方法,为此类化合物的实际合成提供了新思路。
[0024]
2、本发明采用生物质低共熔溶剂作为反应介质,以及采用钴盐作为催化剂,价格低廉,来源易得,环境友好,且低共熔溶剂/钴盐体系可以循环使用6次以上。
[0025]
3、本发明使用的反应底物来源便利,苯甲腈、醇和碳酸亚乙烯酯均为工业化产品,价格十分低廉。官能团兼容性好、底物适用范围广、产率高。
[0026]
4、本发明提供了3,4号位未被取代的1-烷氧基异喹啉化合物的绿色合成方法,可以通过其它有机反应手段在3,4号位引入新的官能团,从而可以实现系列1-烷氧基异喹啉化合物的构建,满足产物多样性。
具体实施方式
[0027]
本发明不局限于下列具体实施方式,本领域一般技术人员根据本发明公开的内容,可以采用其他多种具体实施方式实施本发明的,或者凡是采用本发明的设计结构和思路,做简单变化或更改的,都落入本发明的保护范围。需要说明的是,在不冲突的情况下,本发明中的实施例及实施例中的特征可以相互组合。
[0028]
对照实施例:
[0029]
以下反应式为最佳条件下在低共熔溶剂和钴催化剂作用下发生分子内c-h活化/环化反应(作为标准反应):
[0030][0031]
具体操作步骤为:在一个洁净干燥的25ml圆底烧瓶中依次加入苯甲腈(0.5mmol)、甲醇(10mmol)、碳酸亚乙烯酯(1.5mmol),cp*co(co)i2(5mol%)、醋酸铜(20mol%)、低共熔溶剂甜菜碱/六氟异丙醇(5ml),所得混合液在110℃下加热反应12h,反应结束后,将反应液冷却至室温,乙酸乙酯萃取反应物,用石油醚(pe)/乙酸乙酯(ea)作为洗脱剂,采用硅胶(200 300目筛)进行柱色谱纯化。
[0032]
以下对照实验组1~19是以标准反应条件为参照,进行对比说明:
[0033][0034][0035]
上表中实验组1~5考察了不同催化剂对反应的影响,从实验数据可以看出,三种钴催化剂对反应都具有一定的催化活性,产率均在65%以上,且cp*co(co)i2用量为5mol%时,反应效果最佳为92%。
[0036]
实验组6~9考察了添加剂对反应的影响,其中醋酸铜的的效果最好。
[0037]
实验组10~16考察了不同低共熔溶剂、组成及用量对反应的影响。其中,甜菜碱/六氟异丙醇作为溶剂效果最好。低共熔溶剂中甜菜碱和六氟异丙醇的最佳摩尔比例为1:2,过高或过低都会降低目标产物收率,说明甜菜碱和六氟异丙醇之间存在明显的协同增效作用。另外,甜菜碱/六氟异丙醇的用量对反应也有一定的影响,用量过高不会降低目标产物收率,但是用量过低会在一定程度上降低目标产物收率,最好是保证反应物浓度在0.05~0.1mol/l的浓度范围内。
[0038]
实验组17~19考察了不同反应时间和温度对反应的影响,反应温度最优选在110℃左右,温度进一步升高时,目标产物的收率无增加;降低反应温度时,目标产物的收率降低明显;进一步延长反应时间,目标产物的收率无增加。
[0039]
以下实施例1~6按上述标准反应进行:
[0040][0041]
具体操作步骤为:在一个洁净干燥的25ml圆底烧瓶中依次加入芳腈(0.5mmol)、醇(10mmol)、碳酸亚乙烯酯(1.5mmol),cp*co(co)i2(5mol%)、醋酸铜(20mol%)、低共熔溶剂甜菜碱/六氟异丙醇(5ml),所得混合液在110℃下加热反应12h,反应结束后,将反应液冷却至室温,乙酸乙酯萃取反应物,用石油醚(pe)/乙酸乙酯(ea)作为洗脱剂,采用硅胶(200 300目筛)进行柱色谱纯化。
[0042]
实施例1
[0043]
1-甲氧基异喹啉,产率92%。
[0044]1h nmr(500mhz,cdcl3)δ8.26(ddd,j=0.8,2.0,8.2hz,1h),8.01(d,j=6.2hz,1h),7.73(dt,j=0.8,8.2hz,1h),7.65(ddd,j=1.6,7.0,8.2hz,1h),7.53(ddd,j=1.2,7.0,8.2hz,1h),7.21(dd,j=0.8,5.9hz,1h),4.15(s,3h).
[0045]
13
c nmr(125mhz,cdcl3)δ160.9,139.6,137.8,130.4,126.5,126.0,124.1,119.8,114.9,53.6.
[0046]
实施例2
[0047]
1-烯丙氧基异喹啉,产率86%。
[0048]1h nmr(500mhz,cdcl3)δ8.32(dd,j=1.2,8.2hz,1h),8.00(d,j=5.9hz,1h),7.73(d,j=8.2hz,1h),7.66(ddd,j=1.2,6.6,8.2hz,1h),7.54(ddd,j=1.6,6.9,8.3hz,1h),7.22(d,j=5.5hz,1h),6.23(tdd,j=5.4,10.6,17.2hz,1h),5.51(qd,j=1.6,17.3hz,1h),5.32(qd,j=1.4,10.5hz,1h),5.07(td,j=1.6,5.5hz,2h).
[0049]
13
c nmr(125mhz,cdcl3)δ160.1,139.6,137.9,133.5,130.4,126.5,125.7,124.1,119.7,117.1,115.0,66.7.
[0050]
实施例3
[0051]
1-三氟乙氧基异喹啉,产率84%。
[0052]1h nmr(500mhz,cdcl3)δ8.30(ddd,j=0.8,2.0,8.6hz,1h),7.98(d,j=5.9hz,1h),7.78(d,j=8.2hz,1h),7.71(ddd,j=1.2,6.6,8.2hz,1h),7.60(ddd,j=1.2,6.9,8.3hz,1h),7.32(dd,j=0.8,5.9hz,1h),4.97(q,j=8.6hz,2h).
[0053]
13
c nmr(125mhz,cdcl3)δ158.7,139.3,138.4,131.2,128.3,127.5,126.5,125.5,124.2,122.8.
[0054]
实施例4
[0055]
1-甲氧基-6-甲基异喹啉,产率84%。
[0056]1h nmr(500mhz,cdcl3)δ8.18(d,j=7.5hz,1h),8.09(d,j=7.4hz,1h),7.28
–
7.19(m,2h),7.03(dd,j=7.5,1.1hz,1h),4.06(s,3h),2.50(s,3h).
[0057]
13
c nmr(125mhz,cdcl3)δ158.1,143.6,138.0,137.7,128.6,126.2,125.8,120.5,117.3,54.4,21.7.
[0058]
实施例5
[0059]
1,5-二甲氧基异喹啉,产率87%。
[0060]1h nmr(500mhz,cdcl3)δ8.17(d,j=7.3hz,1h),7.39(d,j=1.4hz,1h),7.27(dd,j=7.5,1.4hz,1h),7.19(dd,j=7.5,1.4hz,1h),6.94(dd,j=7.5,1.5hz,1h),4.06(s,3h),3.81(s,3h).
[0061]
13
c nmr(125mhz,cdcl3)δ159.6,156.2,139.3,132.1,127.9,121.0,119.8,115.6,110.2,56.1,54.4.
[0062]
实施例6
[0063]
5-氯-1-甲氧基异喹啉,产率81%。
[0064]1h nmr(500mhz,cdcl3)δ8.36(d,j=7.5hz,1h),8.15(dd,j=7.5,1.4hz,1h),7.50(d,j=7.5hz,1h),7.41(dd,j=7.5,1.4hz,1h),7.31(t,j=7.5hz,1h),4.06(s,3h).
[0065]
13
c nmr(125mhz,cdcl3)δ156.2,144.5,138.4,133.5,128.7,127.8,124.0,119.8,112.8,54.4.
[0066]
实施例7
[0067]
具体操作步骤为:在一个洁净干燥的25ml圆底烧瓶中依次加入苯甲腈(0.5mmol)、甲醇(10mmol)、碳酸亚乙烯酯(1.5mmol),cp*co(co)i2(5mol%)、醋酸铜(20mol%)、低共熔溶剂甜菜碱/六氟异丙醇(5ml),所得混合液在110℃下加热反应12h,反应结束后,将反应液冷却至室温,乙酸乙酯萃取反应物,核磁分析产物产率。再在萃余液中依次加入新的苯甲腈(0.5mmol)、甲醇(10mmol)、碳酸亚乙烯酯(1.5mmol),反应体系在110℃下加热反应12h,反应完成后,采用乙酸乙酯萃取反应物,核磁分析产物产率。依次重复5次。
[0068]
循环次数123456产率92%91%90%88%86%85%
[0069]
本发明合成的3,4号位未被取代的1-烷氧基异喹啉化合物,可以通过其它有机反应手段在3,4号位引入新的官能团,从而可以实现系列1-烷氧基异喹啉化合物的构建,满足产物多样性,例如以下实施例:
[0070]
实施例8
[0071]
1-甲氧基异喹啉化合物4号位氯代官能团化反应,反应式如下:
[0072][0073]
具体操作步骤为:在一个10ml圆底烧瓶中依次加入1-甲氧基异喹啉(0.4mmol)、乙酰碘苯(0.6mmol)、氯甲酸乙酯(2mmol)、二氯乙烷(2ml),所得混合液在50℃下加热反应6h,反应结束后,将反应液冷却至室温,乙酸乙酯萃取反应物,用石油醚(pe)/乙酸乙酯(ea)作为洗脱剂,采用硅胶(200 300目筛)进行柱色谱纯化。
[0074]
4-氯-1-甲氧基异喹啉,产率85%。
[0075]1h nmr(500mhz,cdcl3)δ8.25(dd,j=7.5,1.4hz,1h),8.08(s,1h),8.02(dd,j=7.5,1.4hz,1h),7.62(td,j=7.5,1.5hz,1h),7.47(td,j=7.5,1.4hz,1h),4.06(s,3h).
[0076]
13
c nmr(125mhz,cdcl3)δ156.5,142.9,131.1,129.7,126.6,126.0,124.3,123.9,123.3,54.4.
[0077]
实施例9
[0078]
1-甲氧基异喹啉化合物3号位氨基化反应,反应式如下:
[0079][0080]
具体操作步骤为:在一个10ml圆底烧瓶中依次加入1-甲氧基异喹啉(0.4mmol)、苯并三氮唑(0.8mmol)、醋酸铜(0.04mmol)、selectfluor(0.6mmol)、碳酸钾(0.8mmol)、硝基甲烷(3ml),所得混合液在120℃下加热反应12h,反应结束后,将反应液冷却至室温,乙酸乙酯萃取反应物,用石油醚(pe)/乙酸乙酯(ea)作为洗脱剂,采用硅胶(200 300目筛)进行柱色谱纯化。
[0081]
3-(1-苯并三氮唑)-1-甲氧基异喹啉,产率78%。
[0082]1h nmr(500mhz,cdcl3)δ8.27(dd,j=7.5,1.2hz,1h),7.97(ddd,j=10.5,9.6,2.6hz,3h),7.64
–
7.52(m,2h),7.51
–
7.34(m,3h),4.06(s,3h).
[0083]
13
c nmr(125mhz,cdcl3)δ152.0,144.5,142.8,137.8,134.3,130.5,129.3,128.2,126.4,126.3,124.8,118.2,116.6,115.3,106.1,54.4.
[0084]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1