一种泰地罗新的合成方法与流程
:
1.本发明属于药物合成技术领域,更具体地涉及一种泰地罗新的合成方法。
背景技术:
2.泰地罗新(tildipirosin)是英特威公司开发的最新动物专用的大环内酯类半合成抗生素,为泰乐菌素的衍生物,对牛、猪的呼吸道疾病具有十分明显的治疗效果,药效强于泰乐菌素、替米考星,而且具备动物专用、用量少、一次给药全程治疗、超长的消除半衰期、生物利用度高、低残留等优点,其化学名为:20,23-二哌啶基-5-氧-碳霉胺糖基-泰乐内酯,2011年3月8日,欧盟兽用药品委员会(cvmp)准许了英特威公司以泰地罗新为主要成分的无菌注射液的市场许可申请,随后在欧盟国家相继批准上市。
3.其结构式如下
[0004][0005]
对于该化合物的合成,目前已经报道的方法主要包括以下几种:
[0006]
专利cn108033988a:泰乐菌素通过胺化反应将泰乐菌素20位的醛基转化为哌啶基,再通过水解反应脱除4位和15位的糖基,然后依次经氧化反应、胺化反应将23位的羟基转化为哌啶基,最终得到泰地罗新。这种方法操作繁琐,步骤多,造成产品收率低,且泰地罗新纯度仅为97%,产品纯度低。
[0007]
专利wo2008012343为原研公司报道的合成方法,以泰乐菌素为原料,经胺化还原、高浓度强酸水解、碘化物活化23位,然后再次进行胺化反应得到泰地罗新。这种方法得到的副产物较多,产品总收率仅为12.2%,不适合工业化生产。
[0008]
专利cn102863487公开了一种新的制备方法,该方法以酒石酸泰乐菌素为起始原料,经水解、胺化、碘代、二次胺化得到20,23-二哌啶基-5-o-碳霉胺糖基-泰乐内酯。该方法仍需经过水解、胺化、碘代、二次胺化得到泰地罗新原料药,步骤复杂、收率较低、后处理繁琐,三废量较高。
[0009]
专利us6514946报道了一种以20,23-二碘-5-o-碳霉胺糖基-泰乐内酯为原料,与哌啶在乙腈中回流反应,反应液经过柱层析,得到泰地罗新的方法,该方法20,23-二碘-5-o-碳霉胺糖基-泰乐内酯不易得,柱层析也不利于工业推广。
[0010]
专利cn201410108841报道一种以酒石酸泰乐菌素为原料,经过氨化,高浓度强酸水解,次氯酸钠氧化,再与哌啶氨化制得泰地罗新的方法,该方法是使23位的羟基反应生成醛基,该反应为选择性氧化反应,人们不易将其反应过程严格控制在生成醛基阶段,并且醛一般都不稳定,所以反应进程难以精确控制。反应条件苛刻,工艺路线控制困难,收率不高。
[0011]
专利cn102863487a报道了一种以酒石酸泰乐菌素为原料的方法,其首先利用氢溴酸对原料进行水解反应,并引入哌啶基团,制备出20-哌啶基-23-羟基-5-o-碳霉胺糖基-泰乐内酯,然后对23位进行碘代反应,最后于23位引入哌啶基团。该方法碘代反应需要用到三苯基膦和单质碘,三苯基膦的副产物毒性较大,容易对环境造成污染;单质碘的价格较高,提高了产品的单价,不利于市场竞争。
[0012]
cn104892704报道:酒石酸泰乐菌素经过水解后,用tempo将23位羟基氧化为醛基,然后经过还原胺化反应得到泰地罗新。tempo氧化性强,这容易造成其他部位的羟基被氧化,从而使产品的纯度很低,工业大生产不可控。
[0013]
综上所述:以上方法都存在或多或少的问题:有的合成步骤多,产品收率低;有的催化剂毒性大、价格高,经济上不可行;有的产品副产物高,造成产品纯度低;有的工业大生产不好控制,产能低。因此开发一种环境友好、经济可行、高纯度的泰地罗新合成方法是当前研究的热点。
技术实现要素:
[0014]
为解决上述问题,克服现有技术的不足,本发明提供了一种泰地罗新的合成方法,减少了二类溶剂的使用,无有害气体的产生,而且反应温度和条件温和易于控制,降低了危险产生的概率,同时该方法成本低、环保,产品的收率高、纯度高。
[0015]
本发明解决上述技术问题的具体技术方案为:一种泰地罗新的合成方法,包括以下工艺步骤:
[0016]
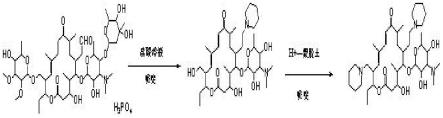
步骤(ⅰ):将起始物料磷酸泰乐菌素悬浮于有机溶媒,并在盐酸溶液的作用下,加入哌啶进行反应,经过调节ph、萃取、干燥和蒸馏得到中间体;
[0017]
步骤(ⅱ):将中间体溶于极性溶剂,在催化剂的作用下,加入哌啶进行反应,经过调节ph、萃取、蒸馏、养晶和抽滤得到泰地罗新。
[0018]
进一步地,所述催化剂为h+-蒙脱土。
[0019]
进一步地,所述步骤(ⅰ)包括:
[0020]
(1)将起始物料磷酸泰乐菌素悬浮于有机溶媒待用:所述的有机溶媒包括乙酸乙酯或乙酸丁酯或乙醚,磷酸泰乐菌素与有机溶媒的质量体积比为1:5~20;
[0021]
(2)将上述磷酸泰乐菌素悬浮液中加入盐酸溶液;所述盐酸溶液的摩尔浓度为1.0~1.8mol/l;磷酸泰乐菌素与盐酸溶液的质量体积比为1:2.0~5.0;
[0022]
(3)反应温度控制在50~77℃,向上述反应液中加入哌啶,反应时间为5~10h;所述的磷酸泰乐菌素与哌啶的用量质量比为1:0.15~0.25;
[0023]
(4)反应结束后,将水加入上述反应液中,利用碱液调节ph至6.5~7.5,分层萃取,取有机相,加入无水硫酸钠干燥,抽滤后蒸馏即得中间体;所述碱液包括碳酸钠或碳酸钾或氨水或三乙胺。
[0024]
进一步地,所述步骤(ⅱ)包括:
[0025]
(1)将中间体加入极性溶剂中溶解待用,所述的极性溶剂包括环丁砜、二甲亚砜、1,4-二氧六环,中间体与极性溶剂的质量体积比为1:6~12;
[0026]
(2)将上述中间体溶解液中加入催化剂h+-蒙脱土;所述的中间体与h+-蒙脱土的质量比为1:0.2~0.5;
[0027]
(3)反应温度控制在60~90℃,向上述反应液中加入哌啶,反应时间为6~8h;所述的中间体与哌啶的用量质量比为1:0.10~0.25;
[0028]
(4)反应结束后,将水和乙酸乙酯加入上述反应液中,利用碱液调节ph至8.0~9.5,分层萃取,取有机相,加入无水硫酸钠干燥,抽滤后蒸馏,降温养晶,抽滤干燥后得到泰地罗新;
[0029]
所述碱液包括碳酸钠或碳酸钾或氨水或三乙胺;所述的养晶温度为-5~5℃。
[0030]
进一步地,所述步骤(ⅰ)中调节ph、萃取、干燥控制温度在20~30℃。
[0031]
进一步地,所述步骤(ⅱ)中调节ph、萃取、干燥控制温度在25~35℃。
[0032]
进一步地,所述中间体为23-羟基-20-哌啶基-5-氧-碳霉胺糖基-泰乐内酯。
[0033]
本发明的有益效果是:
[0034]
提供一种合成泰地罗新的方法,该方法操作简单,反应周期短,且中间体无需进行干燥,可直接投入下一步反应,且无需过柱纯化,有效缩短了制备周期,提高了工作效率,泰地罗新纯度在98.5%以上;
[0035]
极大地减少了二类溶剂的使用,极大的提高了纯度和安全性,并且杂质中的有害物质较少,易于精制处理;具有污染少、环保、工艺简单、条件温和、成本低、产品纯度高等特点,非常适合工业化大生产。
附图说明:
[0036]
附图1是本发明原理图;
具体实施方式:
[0037]
在本发明的描述中具体细节仅仅是为了能够充分理解本发明的实施例,但是作为本领域的技术人员应该知道本发明的实施并不限于这些细节。另外,公知的结构和功能没有被详细的描述或者展示,以避免模糊了本发明实施例的要点。对于本领域的普通技术人员而言,可以具体情况理解上述术语在本发明中的具体含义。
[0038]
本发明的具体实施方式:
[0039]
为了更好的理解本发明特以具体的实施例进行说明,值得强调的是该实施例的效果与本发明保护范围内的各种实施例,包括各自试剂及试剂的含量配比无实质性差异,均能够实现本发明所描述的效果及解决上述问题,其他组合在此不做累述;
[0040]
实施例(1):
[0041]
取磷酸泰乐菌素101.41g(0.1mol)于三口烧瓶中,以650ml有机溶媒乙酸乙酯作为反应溶剂,搅拌条件下加入320ml1.6mol/l盐酸溶液和17.03g(0.2mol)哌啶,升温至65℃,计时反应8h;反应结束后,将100ml水加入上述反应液中,降温至20~30℃;利用饱和碳酸钠溶液调节ph至6.5~7.5,搅拌约10min后分层,弃水相,取有机相;加入无水硫酸钠干燥约30min,抽滤得抽滤母液,蒸馏至液滴基本不再滴出即得中间体54.8g。
[0042]
将中间体溶于900ml的极性溶剂1,4-二氧六环,加入30.42g催化剂h+-蒙脱土以及12.77g哌啶(0.15mol),升温至80℃,计时反应7h;反应结束后,将300ml水和1.5l萃取剂乙酸乙酯加入上述反应液中,利用饱和碳酸钠溶液调节ph至8.0~9.5,分层萃取,取有机相,加入无水硫酸钠干燥,抽滤后蒸馏至体积剩余约200ml,降温至0℃养晶约12h,抽滤,乙酸乙
酯洗涤,干燥后即得泰地罗新49.62g。
[0043]
实施例(2):
[0044]
取磷酸泰乐菌素30.42g(0.03mol)于三口烧瓶中,以200ml有机溶媒乙酸丁酯作为反应溶剂,搅拌条件下加入96ml1.3mol/l盐酸溶液和5.11g(0.06mol)哌啶,升温至65℃,计时反应8h。反应结束后,将30ml水加入上述反应液中,降温至20~30℃。利用饱和碳酸钠溶液调节ph至6.5~7.5,搅拌约10min后分层,弃水相,取有机相,加入无水硫酸钠干燥约30min,抽滤得抽滤母液,蒸馏至液滴基本不再滴出即得中间体16.4g;
[0045]
将中间体溶于270ml的极性溶剂1,4-二氧六环,加入10.14g催化剂h+-蒙脱土以及4.26g哌啶(0.045mol)。升温至80℃,计时反应7h。反应结束后,将100ml水和500ml萃取剂乙酸乙酯加入上述反应液中,利用饱和碳酸钠溶液调节ph至8.0~9.5,分层萃取,取有机相。加入无水硫酸钠干燥,抽滤后蒸馏至体积剩余约60ml,降温至0℃养晶约12h,抽滤,乙酸乙酯洗涤,干燥后即得泰地罗新14.32g;
[0046]
实施例(3):
[0047]
取磷酸泰乐菌素50.71g(0.05mol)于三口烧瓶中,以400ml有机溶媒乙醚作为反应溶剂,搅拌条件下加入175ml1.4mol/l盐酸溶液和8.52g(0.1mol)哌啶,升温至65℃,计时反应8h。反应结束后,将50ml水加入上述反应液中,降温至20~30℃。利用饱和碳酸钠溶液调节ph至6.5~7.5,搅拌约10min后分层,弃水相,取有机相。加入无水硫酸钠干燥约30min,抽滤得抽滤母液,蒸馏至液滴基本不再滴出即得中间体26.21g;
[0048]
将中间体溶于450ml1,4-二氧六环,加入15.21g催化剂h+-蒙脱土以及6.39g哌啶(0.075mol)。升温至80℃,计时反应7h。反应结束后,将150ml水和750ml萃取剂乙酸乙酯加入上述反应液中,利用饱和碳酸钠溶液调节ph至8.0~9.5,分层萃取,取有机相。加入无水硫酸钠干燥,抽滤后蒸馏至体积剩余约100ml,降温至0℃养晶约12h,抽滤,乙酸乙酯洗涤,干燥后即得泰地罗新24.28g;
[0049]
实施例(4):
[0050]
取磷酸泰乐菌素60.84g(0.06mol)于三口烧瓶中,以400ml有机溶媒乙酸乙酯作为反应溶剂,搅拌条件下加入200ml1.3mol/l盐酸溶液和10.22g(0.12mol)哌啶,升温至65℃,计时反应8h。反应结束后,将30ml水加入上述反应液中,降温至20~30℃。利用饱和碳酸钠溶液调节ph至6.5~7.5,搅拌约10min后分层,弃水相,取有机相。加入无水硫酸钠干燥约30min,抽滤得抽滤母液,蒸馏至液滴基本不再滴出即得中间体33.36g;
[0051]
将中间体溶于350ml二甲基亚砜,加入20.28g催化剂h+-蒙脱土以及8.52g哌啶(0.09mol)。升温至80℃,计时反应7h。反应结束后,将200ml水和1000ml萃取剂乙酸乙酯加入上述反应液中,利用饱和碳酸钠溶液调节ph至8.0~9.5,分层萃取,取有机相。加入无水硫酸钠干燥,抽滤后蒸馏至体积剩余约120ml,降温至0℃养晶约12h,抽滤,乙酸乙酯洗涤,干燥后即得泰地罗新29.10g;
[0052]
实施例(5):
[0053]
取磷酸泰乐菌素91.26g(0.09mol)于三口烧瓶中,以600ml的有机溶媒乙酸乙酯作为反应溶剂,搅拌条件下加入300ml1.3mol/l盐酸溶液和15.33g(0.18mol)哌啶,升温至65℃,计时反应8h。反应结束后,将30ml水加入上述反应液中,降温至20~30℃。利用饱和碳酸钠溶液调节ph至6.5~7.5,搅拌约10min后分层,弃水相,取有机相。加入无水硫酸钠干燥约
30min,抽滤得抽滤母液,蒸馏至液滴基本不再滴出即得中间体47.52g;
[0054]
将中间体溶于270ml环丁砜,加入36.50g催化剂h+-蒙脱土以及12.78g哌啶(0.135mol)。升温至80℃,计时反应7h。反应结束后,将300ml水和1500ml萃取剂乙酸乙酯加入上述反应液中,利用饱和碳酸钠溶液调节ph至8.0~9.5,分层萃取,取有机相。加入无水硫酸钠干燥,抽滤后蒸馏至体积剩余约200ml,降温至0℃养晶约12h,抽滤,乙酸乙酯洗涤,干燥后即得泰地罗新42.99g;
[0055]
为了更加直观的展现本发明的工艺优势,特以本发明泰地罗新的合成方法和相同工艺采用替换的方法进行对比,
[0056]
对比例一:
[0057]
制备方法同实施例,所不同的是:本对比例的制备过程中,未添加催化剂为h+-蒙脱土;
[0058]
并根据《中国药典》中关于泰地罗新的检测方法进行检测,试验结果如下:
[0059]
表1:不同实施例和对比例产品属性对比数据
[0060] 中间体的摩尔收率泰地罗新摩尔收率泰地罗新的纯度二类溶剂实施例(1)82.04%82.41%98.8%未检出实施例(2)81.84%79.47%98.9%未检出实施例(3)78.47%84.31%99.1%未检出实施例(4)83.23%79.38%98.7%未检出实施例(5)79.04%82.32%99.2%未检出对比例(1)80.05%12.15%73.7%未检出
[0061]
由上述表格可知:
[0062]
根据实施例1-5和对比例1数据相比:作为中间体的23-羟基-20-哌啶基-5-氧-碳霉胺糖基-泰乐内酯摩尔收率影响不大,而对于泰地罗新的摩尔收率和泰地罗新的纯度影响较大;
[0063]
这可能是由于没有添加催化剂所导致的,中间体的23-羟基-20-哌啶基-5-氧-碳霉胺糖基-泰乐内酯无法很好地与哌啶进行反应,再经过调节ph、萃取、蒸馏、养晶和抽滤得到纯度较高收率较好的泰地罗新。
[0064]
最为重要的是:本发明提供的一种合成泰地罗新的方法,和技术背景中公开的多项对比文献相比,该方法操作简单,反应周期短,且中间体无需进行干燥,可直接投入下一步反应,且无需过柱纯化,有效缩短了制备周期,提高了工作效率,泰地罗新纯度在98.5%以上;
[0065]
与此同时,本发明提供的一种合成泰地罗新的方法,能够极大地减少了二类溶剂的使用,仅需要在中间体溶解时少量引入,极大地减少了二类溶剂对环境的影响;且经过本发明的后续步骤处理后,二类溶剂残留符合国家药典的要求,极大的提高了纯度和安全性,有害物质较少,易于精制处理;具有污染少、环保、工艺简单、条件温和、成本低、产品纯度高等特点,非常适合工业化大生产。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1