一类具有α-葡萄糖苷酶抑制活性的酚类化合物的制备方法与用途
一类具有
α-葡萄糖苷酶抑制活性的酚类化合物的制备方法与用途
技术领域
1.本发明涉及医药技术领域,具体地涉及一类从润楠属植物红梗润楠中分离得到的具有α-葡萄糖苷酶抑制活性的酚类化合物及该类化合物的制备和应用。
背景技术:
2.樟科润楠属植物多为常绿乔木或灌木,全球约有100余种,主要分布于亚洲东南部和东部的热带、亚热带。在我国约68种3变种,主要分布于西南、中南部至台湾,北达山东、湖北及甘肃和陕西南部。根据文献报道,润楠属植物具有丰富多样的次生代谢产物,包括黄酮类、木脂素、倍半萜以及三萜皂苷等。这些化合物多具有显著的生物活性,如抗氧化、与抗炎、神经保护、细胞毒及α-葡萄糖苷酶抑制等生物活性。
3.α-葡萄糖苷酶抑制剂通过竞争性地抑制小肠粘膜刷状缘的α-葡萄糖苷酶,延缓复杂碳水化合物的消化和肠道对葡萄糖的吸收,从而降低餐后血糖水平。α-葡萄糖苷酶抑制剂具有疗效好、药性温和、毒副作用小等优点,但都具有严重的胃肠道副作用,例如腹胀,抽筋,或腹泻等。因此,临床上迫切需要新的更加安全高效的α-葡萄糖苷酶抑制剂。
4.截至目前,国内外还未有学者对红梗润楠的化学成分及药理活性进行研究,因此,对我国境内的红梗润楠进行化学成分研究和α-葡萄糖苷酶抑制活性筛选,阐明其活性成分并进行药物开发具有重大意义。
5.本发明从红梗润楠中提取分离、制备的酚类化合物1-7在α-葡萄糖苷酶抑制剂类药物的制备、2型糖尿病治疗药物的研究具有重要的意义和前景。
技术实现要素:
6.本发明的目的在于提供一类具有α-葡萄糖苷酶抑制活性的酚类化合物的制备及其用途,以解决上述背景技术中提出的问题。
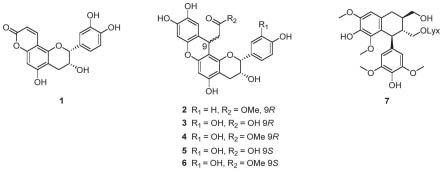
7.为实现上述目的,本发明提供如下技术方案:一类具有α-葡萄糖苷酶抑制活性的酚类化合物,其分别为化合物1至化合物7,所述的化合物1的化学式为:所述的化合物2至化合物6的化学式为:
所述的化合物7的化学式为:
8.其中,化合物2的r1基团为氢,化合物3至化合物6的r1基团为羟基,化合物2和化合物4的r2基团为手性为9r的甲氧基,化合物3的r2基团为手性为9r的羟基,化合物5的r2基团为手性为9s的羟基,化合物6的r2基团为手性为9s的甲氧基。
9.上述化合物1至化合物7的制备步骤如下:
10.步骤一、制备提取物浸膏
11.将干燥的润楠属植物红梗润楠粉碎后用乙醇水溶液按浸渍法提取,得提取液,将提取液减压浓缩回收乙醇,得粗浸膏;
12.步骤二、分离纯化
13.首先将步骤一中得到的粗浸膏分散于水中成混悬液,将混悬液,依次用石油醚、乙酸乙酯和正丁醇萃取,所得乙酸乙酯相和正丁醇相分别浓缩得到乙酸乙酯提取浸膏和正丁醇提取浸膏;
14.再将得到的乙酸乙酯提取浸膏进行mci柱层析,以甲醇/水梯度洗脱,根据tlc显色合并相似流份,得到两个组分,其分别为fr.1和fr.2;
15.将所述的fr.1采用200-300目硅胶柱层析,以二氯甲烷/甲醇体系进行梯度洗脱,得到三个亚组分,其分别为fr.1a至fr.1c;
16.将所述的fr.1c组分用sephadex lh-20以纯乙醇洗脱分为五个亚组分,其分别为fr.1c-1至fr.1c-5;
17.将所述的fr.1c-3组分进一步经硅胶柱层析,以二氯甲烷/甲醇体系,得四个亚组分,其分别为fr.1c-3a至fr.1c-3d;
18.将所述的fr.2组分用mci柱层析,以甲醇/水梯度进行洗脱,合并浓缩后得到四个组分,其分别为fr.2a至fr.2d;
19.将所述的fr.2c组分用200-300目硅胶柱层析,以二氯甲烷/甲醇体系进行梯度洗脱,得到四个亚组分,其分别为fr.2c-1至fr.2c-4;
20.接着将得到的正丁醇提取浸膏用mci柱层析进行初步分离,以甲醇/水梯度进行洗脱,根据tlc显色合并相似流份得到两个组分,其分别为fr.3和fr.4;
21.将所述的fr.3组分用200-300目硅胶柱层析,以二氯甲烷/甲醇体系进行梯度洗脱,得到五个亚组分,其分别为fr.3a至fr.3f;
22.将所述的fr.4组分经sephadex lh-20以纯乙醇洗脱分为五个亚组分其分别为fr.4a至fr.4f;
23.将所述的fr.4c组分用200-300目硅胶柱层析,以二氯甲烷/甲醇体系进行梯度洗脱,得到三个亚组分,其分别为fr.4c-1至fr.4c-3;
24.步骤三、通过步骤二中的组分或者亚组分分别提纯得到化合物1至化合物7。
25.在本技术方案中优选的,在步骤三中,
26.将所述的fr.1c-3a经sephadex lh-20纯化得到化合物1;
27.将所述的fr.1c-4以纯乙醇经sephadex lh-20洗脱纯化得到化合物2和化合物5;
28.将所述的fr.2c-4经过高压液相色谱法精制纯化得到化合物3;
29.将所述的fr.1c-5采用200-300目硅胶柱层析,以二氯甲烷/甲醇体系进行梯度洗脱,得到化合物4;
30.将所述的fr.4c-3依次经sephadex lh-20以纯乙醇洗脱和高压液相色谱法精制纯化得到化合物6;
31.将所述的fr.3c经高压液相色谱法精制纯化得化合物7;
32.在本技术方案中优选的,所述步骤一中所使用的乙醇水溶液中乙醇的体积分数为95%。
33.在本技术方案中优选的,步骤二中制备fr.1a至fr.1c时,采用的二氯甲烷和甲醇的体积比为15∶1-5∶2;制备fr.1c-3a至fr.1c-3d时,采用的二氯甲烷和甲醇的体积比为20∶1-5∶1;制备fr.2a至fr.2d时,采用的甲醇和水的体积比为5∶95-40∶60;制备fr.2c-1至fr.2c-4时,采用的二氯甲烷和甲醇的体积比为14∶1-3∶1;制备fr.3和fr.4时,采用的甲醇和水的体积比为10∶90-100∶0;制备fr.3a至fr.3f时,采用的二氯甲烷和甲醇的体积比为9∶1-3∶1;制备fr.4c-1至fr.4c-3时,采用的二氯甲烷和甲醇的体积比为20∶1-6∶1。
34.在本技术方案中优选的,在制备化合物3时,高压液相色谱法中乙腈和水的体积比为20∶80;在制备化合物4时,二氯甲烷和甲醇的体积比为12∶18∶1;在制备化合物6时,高压液相色谱法中乙腈和水的体积比为17∶83;在制备化合物7时,高压液相色谱法中乙腈和水的体积比为14∶86。
35.需要知道的是,可以将通过上述制造方法制作出的化合物1至化合物7中的任意一个或者多个的混合物,应用至糖尿病药物的制备当中。
36.与现有技术相比,本发明的有益效果是:
37.本发明所提出的化合物2和化合物7均为新的化合物,此前从未进行过公开,并且对化合物1至化合物7进行α-葡萄糖苷酶抑制活性试验,结果表明化合物3、化合物4、化合物5和化合物6有显著的α-葡萄糖苷酶抑制活性,其ic
50
值分别为6.3
±
0.05μm、7.0
±
0.2μm、2.0
±
0.2μm和2.1
±
0.02μm,因此,化合物3、化合物4、化合物5和化合物6可用来制备α-葡萄糖苷酶抑制活性剂,用于制备2型糖尿病治疗药物。
38.本发明通过对红梗润楠进行化学成分研究和α-葡萄糖苷酶抑制活性筛选,阐明其活性成分并进行药物开发具有重大意义,从红梗润楠中提取分离、制备的酚类化合物1至化合物7在α-葡萄糖苷酶抑制剂类药物的制备、2型糖尿病治疗药物的研究具有重要的意义和前景。
附图说明
39.图1为化合物1的1h-nmr谱(cd3od);
40.图2为化合物2的1h-nmr谱(cd3od);
41.图3为化合物2的
13
c-nmr谱(cd3od);
42.图4为化合物2的hsqc谱(cd3od);
43.图5为化合物2的hmbc谱(cd3od);
44.图6为化合物2的1h-1
hcosy谱(cd3od);
45.图7为化合物2的noesy谱(cd3od);
46.图8为化合物2的(+)-hresims谱;
47.图9为化合物3的1h-nmr谱(cd3od);
48.图10为化合物4的1h-nmr谱(cd3od);
49.图11为化合物4的
13
c-nmr谱(cd3od);
50.图12为化合物5的1h-nmr谱(cd3od);
51.图13为化合物6的1h-nmr谱(cd3od);
52.图14为化合物7的1h-nmr谱(cd3od);
53.图15为化合物7的
13
c-nmr谱(cd3od);
54.图16为化合物7的hsqc谱(cd3od);
55.图17为化合物7的hmbc谱(cd3od);
56.图18为化合物7的1h-1
hcosy谱(cd3od);
57.图19为化合物7的noesy谱(cd3od);
58.图20为化合物7的(+)-hresims谱;
59.图21为化合物1至化合物7的化学结构式汇总图。
具体实施方式
60.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
61.需要说明的是,在本发明的描述中,术语“上”、“下”、“前”、“后”、“左”、“右”、“竖直”、“水平”、“顶”、“底”、“内”、“外”等指示的方位或位置关系为基于附图所示的方位或位置关系,仅是为了便于描述本发明和简化描述,并不是指示或暗示所指的装置或元件所必须具有特定的方位、以特定的方位构造和操作,因此不能理解为对本发明的限制。
62.此外,应当理解,为了便于描述,附图中所示出的各个部件的尺寸并不按照实际的比例关系绘制,例如某些层的厚度或宽度可以相对于其他层有所夸大。
63.应注意的是,相似的标号和字母在下面的附图中表示类似项,因此,一旦某一项在一个附图中被定义或说明,则在随后的附图的说明中将不需要再对其进行进一步的具体讨论和描述。
64.本发明提供一种实施例:一类具有α-葡萄糖苷酶抑制活性的酚类化合物,其分别为化合物1至化合物7,化合物1的化学式为:化合物2至化合物6的化
学式为:化合物7的化学式为:
65.其中,
66.化合物2的r1基团为氢,化合物3至化合物6的r1基团为羟基,化合物2和化合物4的r2基团为手性为9r的甲氧基,化合物3的r2基团为手性为9r的羟基,化合物5的r2基团为手性为9s的羟基,化合物6的r2基团为手性为9s的甲氧基。
67.在本实施例中其制备步骤如下:
68.步骤一、制备提取物浸膏
69.将7.0kg干燥的润楠属植物红梗润楠粉碎后,用30l体积浓度为95%的乙醇水溶液在室温的条件下按浸渍法提取,每次浸渍7天,总共浸渍提取3次,合并得到提取液,将提取液在≤45℃的温度下进行减压浓缩回收乙醇,得622.2g的粗浸膏;
70.步骤二、分离纯化
71.首先将步骤一中得到的粗浸膏分散于水中采用超声混悬,得到混悬液,将混悬液,依次用石油醚、乙酸乙酯和正丁醇萃取三次,然后将所得乙酸乙酯相和正丁醇相分别进行合并,减压浓缩回收乙酸乙酯和正丁醇,分别得77.2g的乙酸乙酯提取浸膏和97.9g的正丁醇提取浸膏;
72.再将得到的77.2g的乙酸乙酯提取浸膏进行mci柱层析,以甲醇/水梯度洗脱,根据tlc显色合并相似流份,得到两个组分,其分别为6.9g的fr.1和27.2g的fr.2;
73.将fr.1采用200-300目硅胶柱层析,以二氯甲烷/甲醇体系进行梯度洗脱,得到三个亚组分,其分别为fr.1a至fr.1c;
74.将2.602g的fr.1c组分用sephadex lh-20以纯乙醇洗脱分为五个亚组分,其分别为fr.1c-1至fr.1c-5;
75.将272.2mg的fr.1c-3组分进一步经硅胶柱层析,以二氯甲烷/甲醇体系,得四个亚组分,其分别为fr.1c-3a至fr.1c-3d;
76.将27.2g的fr.2组分用mci柱层析,以甲醇/水梯度进行洗脱,合并浓缩后得到四个组分,其分别为fr.2a至fr.2d;
77.将9.125g的fr.2c组分用200-300目硅胶柱层析,以二氯甲烷/甲醇体系进行梯度洗脱,得到四个亚组分,其分别为fr.2c-1至fr.2c-4;
78.接着将得到的97.9g的正丁醇提取浸膏用mci柱层析进行初步分离,以甲醇/水梯度进行洗脱,根据tlc显色合并相似流份得到两个组分,其分别为25.0g的fr.3和1.5g的fr.4;
79.将25.0g的fr.3组分用200-300目硅胶柱层析,以二氯甲烷/甲醇体系进行梯度洗脱,得到五个亚组分,其分别为fr.3a至fr.3f;
80.将3.2g的fr.3c组分经sephadexlh-20以纯乙醇洗脱分为四个亚组分,其分别为fr.3c-1至fr.3c-4;
81.将1.6g的fr.3c-1组分进一步用200-300目硅胶柱层析,以二氯甲烷/甲醇体系进行梯度洗脱,得到八个亚组分,其分别为fr.3c-1a至fr.3c-1h;
82.将1.5g的fr.4组分经sephadexlh-20以纯乙醇洗脱分为五个亚组分其分别为fr.4a至fr.4f;
83.将327.6mg的fr.4c组分用200-300目硅胶柱层析,以二氯甲烷/甲醇体系进行梯度洗脱,得到三个亚组分,其分别为fr.4c-1至fr.4c-3;
84.步骤三、将fr.1c-3a经sephadex lh-20纯化得到化合物1;
85.将fr.1c-4以纯乙醇经sephadex lh-20洗脱纯化得到化合物2和化合物5;
86.将fr.2c-4经过高压液相色谱法精制纯化得到化合物3;
87.将fr.1c-5采用200-300目硅胶柱层析,以二氯甲烷/甲醇体系进行梯度洗脱,得到化合物4;
88.将fr.4c-3依次经sephadex lh-20以纯乙醇洗脱和高压液相色谱法精制纯化得到化合物6;
89.将fr.3c经高压液相色谱法精制纯化得化合物7。
90.在本实施例中,需要知道的是,步骤二中制备fr.1a至fr.1c时,采用的二氯甲烷和甲醇的体积比为15∶1-5∶2;制备fr.1c-3a至fr.1c-3d时,采用的二氯甲烷和甲醇的体积比为20∶1-5∶1;制备fr.2a至fr.2d时,采用的甲醇和水的体积比为5∶95-40∶60;制备fr.2c-1至fr.2c-4时,采用的二氯甲烷和甲醇的体积比为14∶1-3∶1;制备fr.3和fr.4时,采用的甲醇和水的体积比为10∶90-100∶0;制备fr.3a至fr.3f时,采用的二氯甲烷和甲醇的体积比为9∶1-3∶1;制备fr.4c-1至fr.4c-3时,采用的二氯甲烷和甲醇的体积比为20∶1-6∶1。
91.在本实施例中还需要知道的是,在制备化合物3时,高压液相色谱法中乙腈和水的体积比为20∶80;在制备化合物4时,二氯甲烷和甲醇的体积比为12∶1-8∶1;在制备化合物6时,高压液相色谱法中乙腈和水的体积比为17∶83;在制备化合物7时,高压液相色谱法中乙腈和水的体积比为14∶86。
92.按常规经nmr、hresims、ecd和uv等多种现代光谱技术,分别确定了化合物2和化合物7的理化性质,其理化性质如下:
93.化合物2:红色固体。uvλ
max
(meoh):215(3.38),271(2.86),350(2.78)nm;1h-nmr(400mhz)和
13
c-nmr(100mhz)数据,见表2;(+)-hresims m/z 467.1319[m+h]
+
,(calcd for c
25h22
o9h
+
,467.1337)。
[0094]
化合物7:淡黄色油状物。uvλ
max
(meoh):225(3.23),281(1.70)nm;1h-nmr(400mhz)和
13
c-nmr(100mhz)数据,见表3;(+)-hresims m/z 575.2100[m+na]+,(calcd for c
27h36o12
na
+
,575.2099)。
[0095]
本实施例中提供了一种化合物1至化合物7的α-葡萄糖苷酶活性抑制试验:
[0096]
将对硝基苯酚α-d-吡喃葡萄糖苷(p-npg)通过α-葡萄糖苷酶(agi)催化水解产生对硝基苯酚(p-np)和葡萄糖,p-np在205nm处有最大吸收,加入待测样品后,可采取测定吸光值的变化来计算样品对agi的抑制率,并通过与阳性对照药阿卡波糖的ic
50
值进行对比,从而评价样品的α-葡萄糖苷酶活性抑制作用。本试验在96孔培养板中进行,总反应体系为200μl。首先每孔加入98μl pbs缓冲溶液,然后加入不同浓度的阿卡波糖溶液或待测样品2μ
l,再加入25μlα-糖苷酶溶液,轻微震荡混匀,将96孔培养板放置于37℃水浴锅中孵育20min,而后加入25μl p-npg继续孵育15min,孵育结束后,取出96孔培养板,迅速在每孔中加入50μl na2co3溶液终止反应,并在波长205nm处测定每孔吸光值,记录数据,平行测定三次。本试验以pbs溶液替换酶溶液的为空白组,以dmso溶液替换酶溶液的为阴性对照组,其它试剂与样品实验组保持一致。用酶标仪测定在205nm处的吸光度,计算抑制率及ic
50
值。对比测得的待测样品ic
50
值及阳性对照药阿卡波糖的ic
50
值。
[0097]
实验结果:红梗润楠中分离到的七个单体化合物首先在80μm的初级浓度下进行α-糖苷酶抑制率测试,仅对抑制率>50%的化合物进一步通过上述检测方法,以抑制率对抑制剂浓度作图,根据抑制曲线求得七个化合物的ic
50
值,结果如表1所示。
[0098]
表1
[0099]
项目ic
50
(μm)化合物121.6
±
2.3化合物210.7
±
0.7化合物36.3
±
0.05化合物47.0
±
0.2化合物52.0
±
0.2化合物62.1
±
0.02化合物717.8
±
1.8阿卡波糖270.8
±
32.5
[0100]
从表1中测定结果可知,从红梗润楠中发现七个高活性的α-葡萄糖苷酶抑制剂。其中化合物5和6表现出最强的抑制活性,ic
50
值分别为2.0
±
0.2μm和2.1
±
0.02μm,活性是标准对照品阿卡波糖(ic
50
270.8
±
32.5μm)的100多倍,化合物3和化合物4也具有良好的α-葡萄糖苷酶抑制活性,ic
50
值分别为6.3
±
0.05μm和7.0
±
0.2μm。根据上述试验结果可知如下构-效关系:二氢黄烷醇b环上的邻二酚羟基有利于活性增强;c-11位的甲酯基对活性影响不大;c-9位构型对活性也有影响,s构型相较于r构型活性更好。
[0101]
在本实施例中化合物2和化合物7的碳谱、氢谱数据分别如表2至表3所示:
[0102]
表2
[0103][0104]
表3
[0105][0106]
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1